![]()
C.
COMMERCIO O SOMMINISTRAZIONE DI MEDICINALI GUASTI
(art. 443 c.p.)
DATI MEDICO-SCIENTIFICI IN MERITO AL RADICALE DIFETTO DELLA SICUREZZA NONCHÉ IN MERITO ALL’EVIDENTE NATURA SPERIMENTALE NOTI SIN DALL’ATTO DELL’INTRODUZIONE DELL’OBBLIGO VACCINALE
C.1.
Natura sperimentale confermata esplicitamente nei contratti quadro di acquisto (APA) firmati dalla Commissione Europea e dalla Repubblica Italiana
Pubblicazione del contratto quadro di compravendita tra Commissione CE e Pfizer/BioNTech
Come già sopra evidenziato, ormai è stato reso pubblico e pubblicato anche sul sito della RAI il contratto quadro di compravendita stipulato tra la Commissione CE e la Pfizer/BioNTech leggibile in tutte le parti (doc. 4).
Dal contratto, firmato in data 20.11.2020 dalla Commissaria Europea per la Salute Stella Kyriakides e la Presidente del reparto vaccini di Pfizer Biopharmaceuticals Group (che ha firmato per Pfizer e BioNTech), risulta dalla parte I. (Special Conditions) al punto I.12 (Indemnification) che gli stati membri dell’UE (dunque, anche l’Italia) devono tenere indenni i produttore da richieste di risarcimento danni per danni di ogni genere derivanti dall’uso del “vaccino”. Dall’allegato I (Annex I: Vaccine Order Form) risulta al punto 4, testualmente quanto segue:
“4. The Partecipating Member State acknowledges that the Vaccine and materials related to the Vaccine, and their components and constituent materials are being rapidly developed due to the emergency circumstances of the COVID-19 pandemic and will continue to be studied after provision of the Vaccine to the Participating Member States under the APA. The Participating Member State further acknowledges that the long-term effects and efficacy of the Vaccine are not currently known and that there may be adverse effects of the Vaccine that are not currently known. Further, to the extent applicable, the Participating Member State acknowledges that the Vaccine shall not be serialized.”
Traduzione in lingua italiana:
“4. Il partecipante Stato Membro riconosce che il vaccino e i materiali relativi al vaccino, e i loro componenti e materiali vengono sviluppati rapidamente a causa dell’emergenza Covid-19 e continueranno ad essere studiati dopo la fornitura nell’ambito dell’APA del vaccino allo Stato Membro. Lo Stato Membro Partecipante, inoltre, riconosce che gli effetti a lungo termine e l’efficacia del vaccino allo stato non sono noti e che ci possono essere degli effetti avversi del vaccino che allo stato non sono noti.
Dato che il produttore un (1) mese prima (20.11.2020) dell’immissione sul mercato del “vaccino” Comirnaty di Pfizer/BioNTech (21.12.2020) nel contratto ha esplicitamente dichiarato
- che non conosce l’efficacia e non conosce le conseguenze (effetti collaterali) che l’inoculo del “vaccino” può avere e, che per questo motivo, richiedeva di essere tenuto indenne dal rispettivo Stato Membro UE partecipante all’APA (e così anche dall’Italia) per le richieste di risarcimento per danni causati dal vaccino, e
- che avrebbe continuato a studiare l’efficacia e le conseguenze in termini di eventi avversi del “vaccino” dopo la fornitura del “vaccino” al rispettivo Stato Membro UE partecipante all’APA,
non ci può essere alcun minimo dubbio che la sostanza che è stata inoculata ai cittadini dell’Unione Europea (tra cui quelli italiani) come “vaccino”-Covid-19, è ovviamente una sostanza sperimentale! Ogni altra conclusione è una tanto plateale quanto criminale presa in giro di oltre 400 milioni cittadini europei!
I cosiddetti “vaccini”-Covid-19 hanno una palese natura sperimentale!
La clamorosa erroneità delle sentenze della Corte Costituzionale confermata anche dal contenuto dei contratti stipulati dalla Commissione Europea e dalla Repubblica Italiana con i produttori dei cosiddetti “vaccini”-Covid-19
C.2.
Conferma nelle Decisioni di Esecuzione della Commissione Europea per l’immissione sul mercato della mancanza di fondamentali studi sulla sicurezza dei cosiddetti “vaccini”-Covid-19
I cosiddetti “vaccini”-Covid-19 erano stati autorizzati ai sensi del Regolamento 507/2006 solo in via condizionata (perché mancavano fondamentali studi clinici per la conferma dell’efficacia e della sicurezza) per l’immissione sul mercato.
I rispettivi studi, peraltro, non sono mai stati effettuati, come risulta dalle azioni di annullamento delle autorizzazioni presentate in Tribunale dell’UE (doc. 24 e 25).
Che l’autorizzazione a novembre 2021 (all’atto della presentazione della denuncia penale) e dunque anche dopo l’introduzione dell’obbligo vaccinale, fosse di natura “condizionata” alla conferma dell’efficacia e sicurezza con la conduzione di studi clinici, risulta dalle stesse Decisioni di Esecuzione della Commissione Europea.
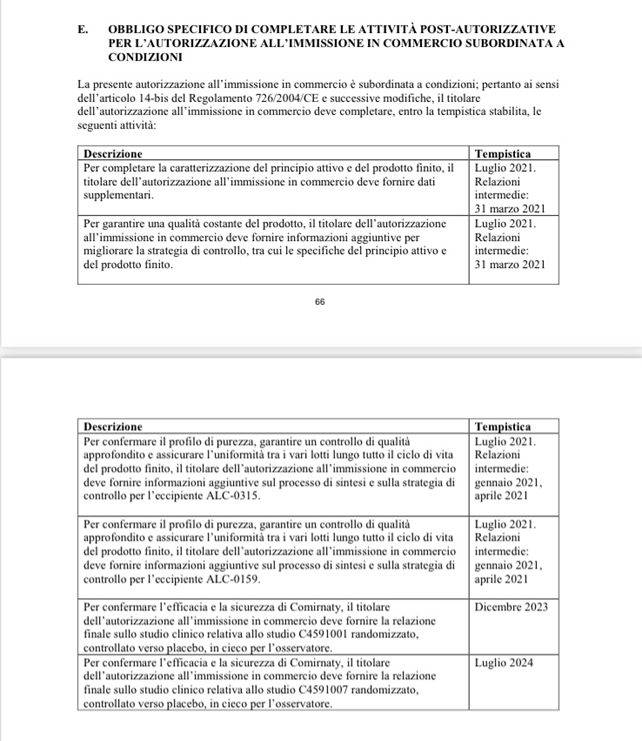
Vedi pag. 18 la Decisione di Esecuzione della Commissione Europea Allegato d.d. 21.12.2020 (già sub doc. 5 Denuncia Penale), laddove dalla documentazione istituzionale della Commissione Europea risulta al riguardo testualmente quanto segue:
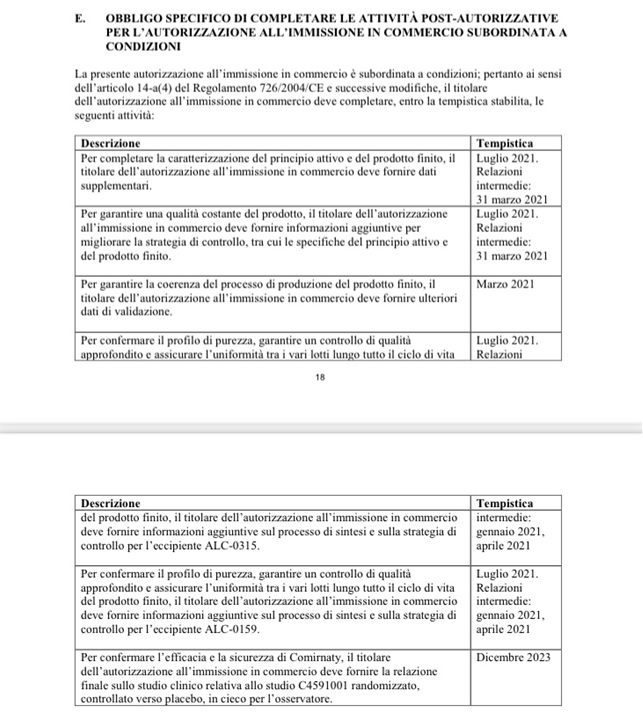
Vedi a pagg. 66 e 67 la Decisione di Esecuzione della Commissione Europea in vigore a novembre 2021
https://ec.europa.eu/health/documents/community-register/2021/20211126154181/anx_154181_it.pdf
(data di presentazione della denuncia e periodo post introduzione dell’obbligo “vaccinale”-Covid-19 – doc. 23), laddove dalla documentazione istituzionale della Commissione Europea risulta al riguardo testualmente quanto segue:
Dalla documentazione istituzionale della Commissione Europea (Allegati I alle rispettive Decisioni di Esecuzione per l’immissione sul mercato dei “vaccini”-Covid-19 risultava sin dall’inizio e risulta a tutt’oggi che mai sono stati fatti – e neanche disposti (!) gli studi di tossicologia e cancerogenicità su queste sostanze NUOVE (come conferma la stessa Commissione Europea nelle sue Decisioni di autorizzazione condizionata).

Come verrà dimostrato infra non sono stati fatti i necessari studi di mutagenicità, e cioè sul potenziale di modifica del genoma umano, e ciò, nonostante che la natura e il meccanismo di queste sostanze lo avesse imposto.
E, intanto, i casi osservati di turbo-cancro e le malattie tumorali, soprattutto tra i giovani, stanno aumentando vertiginosamente in tutto il mondo, e sempre più scienziati – anche tra quelli che inizialmente si erano espressi a favore di questa “vaccinazione” – vanno in pubblico per segnalare il rischio concreto di alterazione del DNA, cioè del genoma umano, che può avere effetti catastrofici ancora imprevedibili nelle loro dimensioni.
Non per niente esiste un divieto di modifica del genoma umano, ancorato anche a livello delle Nazioni Unite!
Vedi la traduzione in lingua italiana dell’azione di annullamento dell’autorizzazione di Comirnaty di Pfizer/BioNTech in Tribunale UE con T-109/23, qui sub doc. 24, l’azione di annullamento di autorizzazione di Spikevax di Moderna in Tribunale UE – T-108/23 (doc. 25), nonché il rispettivo sunto pubblicato anche in lingua italiana in Gazzetta Ufficiale dell’Unione Europea sub doc. 26.1 e 26.2.
Residui di plasmidi (DNA estraneo) nei cosiddetti “vaccini” Covid-19 sono stati rilevati in queste sostanze da diversi laboratori in tutto il mondo, e inoltre è noto dagli anni ’70 che l’RNA può retro-trascriversi nel DNA.
Intanto anche i media nazionali ne parlano (LaVerità, Cresce l’allarme sul DNA nei vaccini: “I residui si concentrano nelle ovaie … Si rischia di colpire geni soppressori del cancro. Necessari controlli a campione… Più tumori nei giovani”. 24.09.2023)
https://drive.google.com/file/d/1nahb1cBGsWJ3CiLq91AKCkfDLZaelyGl/view?usp=drivesdk
(anche in stampa pdf sub doc. 27).
La situazione è molto drammatica e, pertanto la Florida chiede un immediato stop dell’autorizzazione dei “vaccini” a mRNA. Lo scorso 6 dicembre il Dipartimento della Salute del Florida ha inviato una lettera alla Food and drug administration (FDA) degli Stati Uniti e al Centro per il controllo e la prevenzione delle malattie (CDC), dopo la scoperta di miliardi di frammenti di DNA per dose nei vaccini a mRNA di Pfizer e Moderna e il conseguente rischio unico ed elevato per la salute umana e per l’integrità del genoma umano, compreso il rischio che il DNA integrato negli spermatozoi o nei gameti delle uova possa essere tramesso alla prole dei soggetti vaccinati con mRNA contro il Covid-19 (vedi LaVerità, La Florida chiede di abolire i vaccini mRna d.d. 05.01.2024 doc. 28.1 e la lettera del Capo del Dipartimento della Sanità del Florida alla FDA e al CDC sub doc. 28.2.
C.3
Conferma da parte degli stessi produttori nei loro RISK MANAGEMENT PLAN della mancanza di dati fondamentali in punto sicurezza delle sostanze
Nell’ambito della procedura per l’autorizzazione condizionata di immissione sul mercato, i produttori dei cosiddetti “vaccini”-Covid-19 hanno dovuto depositare i cosiddetti Risk Management Plan (piani di gestione dei rischi della somministrazione delle sostanze – RMP). Da quella documentazione istituzionale accettata e pubblicata dall’EMA (Autorità del Farmaco Europea) sul proprio sito web sin dalla immissione sul mercato del rispettivo “vaccino”-Covid-19, risulta in modo inequivocabile che mancano dei/delle fondamentali dati/informazioni sugli effetti e rischi connessi alla somministrazione di queste sostanze. Vedi il Risk Management Plan per “Comirnaty vaccino Covid-19” di Pfizer/BioNTech”- (doc. 6.1 e segg.).
Dal piano di gestione dei rischi presentato dal produttore della sostanza risulta in modo inequivocabile, che ancora addesso nulla si sa in merito ai rischi a medio e lungo termine, in merito all’uso in stato di gravidanza e durante l’allattamento. Non si conoscono gli effetti e rischi che la somministrazione provoca su persone con un problema nel sistema immunitario oppure che hanno in generale un problema di natura infiammatoria nel corpo, circostanze che, peraltro, possono riguardare la stragrande parte dei cittadini! Il produttore, praticamente dichiara che allo stato non sa niente!
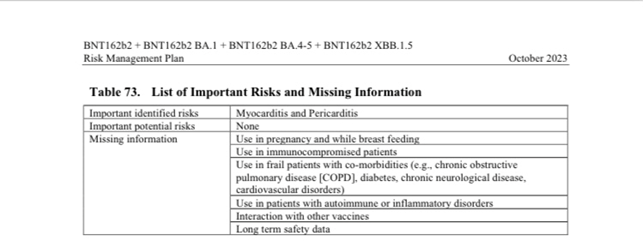
Vedi a pagg. 180 e segg. “Presentation of the Missing Information: use in pregnancy and while breast feeding, Use in frail patients with co-morbidities …, diabetes, chronic neurological disease, cardiovascular disorders, use in patients with autoimmune or inflammatory disorders, interaction with other vaccines, long term safety data.”
C.4
Palese natura sperimentale dei “vaccini”-Covid-19
La natura sperimentale di queste sostanze è palese, non soltanto in punto efficacia ma anche in punto sicurezza. Sono le stesse indicazioni degli effetti collaterali, come indicate nell’Allegato I alle decisioni della Commissione CE di autorizzazione di queste sostanze, che nel loro sviluppo temporale dimostrano la incontestabile natura sperimentale di queste sostanze.
Se all’inizio erano pochi e di lieve entità, ormai la lista degli effetti collaterali diventa sempre più lunga e comprende anche la morte (causata dalla miocardite e pericardite). Ci sono però tantissimi altri effetti collaterali irreversibili che ancora non sono neanche indicati nel foglietto illustrativo.
(vedi doc. 29.1., 29.2. e 29.3).
C.4.1.
Indicazione ufficiale istituzionale della morte quale effetto collaterale dei cosiddetti “vaccini”-covid-19
Da settembre 2023 sopravvenuta indicazione ufficiale, da parte dell’EMA, della Commissione Europea e dell’AIFA, della morte tra gli effetti collaterali
A causa dell’enorme numero di decessi e di altri gravi effetti collaterali e irreversibili dei cosiddetti “vaccini”-Covid-19, l’EMA e la Commissione Europea – che hanno agito, comunque, in modo criminale (vedi la traduzione in lingua italiana dell’azione di annullamento ex art. 263 TFUE in Tribunale UE del 22.02.2023 riguardante il cosiddetto “vaccino”-Covid-19 Comirnaty di Pfizer/BioNTech, doc. 24) hanno dovuto finalmente ammettere pubblicamente e in modo istituzionale che queste iniezioni sperimentali a base genica possono provocare la morte. Lo sapevano però sin dall’inizio, come risulta anche dalla corrispondenza email dei responsabili dell’AIFA con il Ministero della Salute e con i responsabili della autorità sanitarie delle Regioni e Province, resi pubblici dalla redazione di Fuori dal Coro (di Mario Giordano), Rete4, Mediaset. (vedi infra sub C.5.1).
Qui il nuovo punto 4.4. dell’allegato I alla decisione della Commissione Europea di autorizzazione di immissione sul mercato del cosiddetto “vaccino”-Covid-19 Comirnaty di Pfizer/BioNTech

E qui il documento completo:
https://ec.europa.eu/health/documents/community-register/2023/20230831160389/anx_160389_it.pdf
(e in stampa pdf. sub doc. 8).
E qui il documento analogo pubblicato dall’AIFA con decorrenza settembre 2023:
https://www.aifa.gov.it/documents/20142/1279946/RCP_COMIRNATY_XBB.pdf
(e in stampa pdf. sub doc. 30).
Analogo è stato pubblicato da EMA/Commissione Europea per Spikevax di Moderna.

E qui il documento completo:
https://ec.europa.eu/health/documents/community-register/2023/20230915160561/anx_160561_it.pdf
(e in stampa pdf sub doc. 9).
E qui il documento pubblicato dall’AIFA con decorrenza settembre 2023 per Spikevax di Moderna:
(e in stampa pdf sub doc. 31)
Peraltro, i dati ufficiali sono estremamente sottostimati, perché non esiste una farmacovigilanza attiva. Anche nella Provincia Autonoma di Bolzano e in Italia, in generale, in caso di morti improvvise e inaspettate, la “vaccinazione” Covid 19 solitamente non viene considerata dalle autorità come possibile causa… anzi! Ai cittadini viene sistematicamente dichiarato che sarebbe esclusa ogni correlazione!
I dati statistici dell’UE (EUROSTAT) mostrano, invece, una chiara correlazione tra il tasso di mortalità in eccesso e il tasso di “vaccinazione” Covid 19 della popolazione.
I paesi con un alto “tasso di vaccinazione” Covid 19 hanno un tasso di mortalità in eccesso significativamente alto!
C.4.2.
Brutale violazione da parte della Commissione Europea e dell’EMA del diritto del farmaco eurounionale nell’autorizzazione di immissione sul mercato dei cosiddetti “vaccini”-Covid-19
Anche dall’attuale versione degli allegati alle decisioni della Commissione Europea di autorizzazione dell’immissione sul mercato dei cosiddetti “vaccini”-Covid-19 risulta espressamente che non sono stati fatti studi per poter escludere il rischio della genotossicità e cancerogenicità! Sub doc. 8 a titolo esemplificativo la rispettiva documentazione dell’EMA per Comirnaty di Pfizer/BioNTech, laddove risulta testualmente:
“Genotossicità/Potenziale cancerogeno
Non sono stati condotti studi di genotossicità o sul potenziale cancerogeno. Si ritiene che i componenti del vaccino (lipidi e mRNA) non presentino alcun potenziale genotossico.”
Analogo vale per Spikevax di Moderna (vedi sotto www.human.medicinal.register).
La pazzia e criminalità dell’operato dell’EMA (addivengono ad una applicazione in massa su persone sane, “ritenendo” di poter escludere, senza aver fatto alcuno studio invece imposto per legge quale condicio sine qua non!) sono esposte e documentate sia dal punto di vista del diritto farmacologico eurounionale, sia dal punto di vista medico-scientifico, nelle due azioni di annullamento delle autorizzazioni di immissione sul mercato di Comirnaty di Pfizer/BioNTech nonché di Spikevax di Moderna, presentate in Tribunale dell’UE ex art. 263 TFUE dalla sottoscritta difensore per un cittadino italiano, padre di due minorenni (doc. 24 e doc. 25, e sub doc. 26.1. e doc. 26.2. il sunto dei motivi dei due ricorsi pubblicato anche in lingua italiana sulla Gazzetta Ufficiale dell’Unione Europea).
Una delle due sostanze (Comirnaty di Pfizer/BioNTech oppure Spikevax di Moderna) sarebbe stata inoculata ai sanitari denuncianti, dato che Vaxrevia di AstraZeneca e Janssen di Johnson & Johnson non erano mai destinati ai sanitari e nel frattempo sono di fatto stati tolti dalla campagna “vaccinale” in Italia.
A queste due azioni presentate in Tribunale UE sono stati allegati il parere scientifico di Prof.M. Palmer et al, dal quale risultano i reali rischi connessi con l’inoculazione dei cosiddetti “vaccini”-Covid-19 (doc. 32).
I motivi che devono portare all’immediato annullamento delle autorizzazioni per l’immissione sul mercato sono indicati nelle azioni di annullamento scritte in lingua tedesca in modo riassuntivo testualmente come segue (qui tradotto in lingua italiana):”
I° MOTIVO DI NULLITÁ
Gravissima violazione degli artt. 168 e 169 del TFUE e degli artt. 3, 35 e 38 della Carta UE.
della Direttiva 2001/83/CE artt. 8, 11, 26, 54, 58, 59, 86 e seguenti, 101 e seguenti, Allegato I, Parte I, Parte III, Parte IV, nonché del Regolamento (CE) n. 726/2004 artt. 3-7, 10a, 12, 14-a, nonché della Dichiarazione delle Nazioni Unite sul genoma umano e i diritti umani.
aggirando gli elevati standard di sperimentazione previsti per i farmaci a base i ingegneria genetica
- sulla base di un’esclusione infondata e di fatto illogica dell’applicazione delle disposizioni in materia di autorizzazione all’immissione in commercio dei medicinali per terapie avanzate a sostanze che sono dichiarate in termini giuridici puramente formali come vaccini contro le malattie infettive, ma che in realtà corrispondono a medicinali per la terapia genica
- in ogni caso, a causa del mancato coinvolgimento dello specifico Comitato EMA per le Terapie Avanzate, che è necessario, a prescindere dalla classificazione come medicinale di terapia genica, solo sulla base del corredo genetico e della modalità d’azione della sostanza.
- in ogni caso a causa della violazione dei requisiti di autorizzazione per i vaccini basati sull’ingegneria genetica
(1)
La Direttiva 2009/120/UE della Commissione, del 14 settembre 2009, ha modificato la Direttiva 2001/83/UE del Parlamento e del Consiglio, stabilendo che le sostanze dichiarate come vaccini contro le malattie infettive non sono considerate prodotti di terapia genica.
Il testo integrale della DIRETTIVA 2009/120/CE DELLA COMMISSIONE del 14 settembre 2009 che modifica, per quanto riguarda i medicinali per terapie avanzate, la direttiva 2001/83/CE del Parlamento europeo e del Consiglio recante un codice comunitario relativo ai medicinali per uso umano, ALLEGATO “PARTE IV MEDICINALI PER TERAPIE AVANZATE” recita come segue:
2.1. Medicinale di terapia genica
Per medicinale di terapia genica si intende un medicinale biologico che presenta le seguenti caratteristiche:
- Contiene una sostanza attiva che contiene a sua volta o consiste in un acido nucleico ricombinante usato sugli esseri umani o ad essi somministrati per regolare, riparare, sostituire, aggiungere o eliminare una sequenza di acido nucleico.
(b) il suo effetto terapeutico, profilattico o diagnostico è direttamente correlato alla sequenza di acido nucleico ricombinante in esso contenuta o al prodotto dell’espressione genetica di tale sequenza.
I vaccini contro le malattie infettive non sono farmaci di terapia genica.
Questa frase formulata in modo assoluto “i vaccini contro le malattie infettive non sono medicinali di terapia genica” porta al fatto che, a prescindere dalla loro composizione e dalla loro modalità d’azione, le sostanze, per il solo fatto di essere definite “vaccini contro le malattie infettive”, sono semplicemente escluse dalla regolamentazione molto più rigorosa ed esigente dei requisiti di approvazione, che è necessariamente imposta dal legislatore ai medicinali di terapia genica e ai medicinali di terapia avanzata nel loro complesso.
Questo porta all’assurdo che sostanze, pur essendo costruite e agendo come medicinali per la terapia genica, sono escluse dal rigoroso regime di autorizzazione dell’UE per i medicinali per terapie avanzate, necessario per proteggere la salute di tutta la popolazione dell’UE, solo perché sono definite “vaccini contro le malattie infettive”, e sono trattate come i vaccini convenzionali, con i quali non hanno nulla in comune!
Un “vaccino” convenzionale contiene un antigene.
COMIRNATY non contiene antigeni, ma il progetto di parti del virus (proteina spike del virus SARS-CoV-2, che è una pericolosa tossina), e quindi di una sostanza estranea, che l’organismo dovrebbe produrre da solo. Il COMIRNATY è quindi un cosiddetto pro-farmaco.
Pertanto, l’iniezione provoca direttamente la produzione di una sostanza nociva da parte dell’organismo e non di una specifica sostanza di difesa o di protezione, come nel caso delle vaccinazioni convenzionali. La formazione di anticorpi e quindi di sostanze protettive avviene solo nella seconda fase.
È assolutamente incomprensibile il motivo per cui le sostanze contenenti o costituite da un acido nucleico ricombinante che viene iniettato nell’uomo per aggiungere una sequenza di acido nucleico (nel caso specifico, l’mRNA che dovrebbe poi portare alla produzione della proteina spike del SARS Cov-2) siano escluse dalla definizione di “medicinale di terapia genica” e quindi dalle norme di autorizzazione, necessariamente molto severe, per i “medicinali per terapie avanzate”.
A meno che, nel 2009, in modo del tutto deliberato e in violazione dei principi fondamentali del diritto farmaceutico – e quindi del principio di prudenza e del diritto fondamentale alla vita e alla salute, sancito anche dal diritto dell’UE – non si sia creato il presupposto per cui le sostanze che di fatto agiscono come terapie geniche possano essere autorizzate senza rispettare i rigorosi requisiti di autorizzazione che sono necessariamente richiesti per le terapie geniche. Questo sembra essere proprio il caso.
“L’approvazione della terapia genica come vaccinazione convenzionale si è basata su una base scientifica e medico-legale non valida. Questo porta a conseguenze incalcolabili per la salute della popolazione, fino ai bambini più piccoli, alla quale vengono ripetutamente iniettate queste sostanze nelle campagne di “vaccinazione” di massa.
I farmaci a base genica destinati a pochi pazienti con quadri clinici molto specifici sono soggetti a standard di sperimentazione elevati – ma assurdamente non quei farmaci a base genica che sono dichiarati “formalmente” come “vaccini per malattie infettive” (come il COMIRNATY) e vengono iniettati in persone sane (!). Dalla fine di dicembre 2020 al 2 dicembre 2022, quasi un miliardo di dosi di questi “vaccini” sono state somministrate a persone nell’UE – fino all’ottobre 2022 sulla base di autorizzazioni all’immissione in commercio solo condizionate, che poi sono state convertite sic et simpliciter in autorizzazioni all’immissione in commercio non condizionate senza che i produttori avessero soddisfatto le condizioni (vedi sotto), e dunque, in assoluta violazione del diritto dell’UE.
Ciò è avvenuto grazie all’influenza di potenti lobby: con la Direttiva 2009/120/CE, come spiegato in precedenza, la Commissione Europea ha escluso già nel 2009, senza il coinvolgimento del Parlamento Europeo, i “vaccini contro le malattie infettive” dal gruppo di farmaci per la terapia genica appositamente regolamentati, attraverso una ridefinizione legale: “i vaccini contro le malattie infettive non sono farmaci per la terapia genica“. Questa definizione è stata modificata solo in seguito ai commenti dell’industria farmaceutica (doc. A.12). La bozza di direttiva originale (doc. A.13) prevedeva un’ampia definizione di medicinale di terapia genica a favore della tutela della salute pubblica, che avrebbe incluso le iniezioni geniche di Covid 19.
Le aziende farmaceutiche sostenevano, tra l’altro, che i rigidi requisiti di sicurezza previsti dalla bozza di direttiva avrebbero reso la produzione di terapie geniche a base di mRNA notevolmente più costosa. La Commissione europea ha successivamente modificato il testo della direttiva (doc. A.12).
L’esclusione dei vaccini basati su geni contro le malattie infettive dal gruppo dei farmaci per la terapia genica consente ai produttori di risparmiare numerosi studi preclinici, costosi in termini di tempo e denaro. Questi sono essenziali per valutare la sicurezza del farmaco e delle persone che partecipano agli studi clinici.
Le sperimentazioni cliniche non possono essere avviate senza i risultati degli studi preclinici. Di norma, essi fanno luce, tra l’altro, sulla distribuzione dei medicinali nell’organismo, sulla conversione e sulla degradazione biochimica e sulla loro escrezione nell’ambito della cosiddetta farmacocinetica – compreso, nel caso dei medicinali per la terapia genica, il rischio di trasferimento del gene nella linea germinale -, sulle possibili alterazioni del materiale genetico delle cellule (genotossicità), sui rischi di cancro, sull’influenza dei medicinali su parametri importanti per le funzioni di base del corpo umano (farmacologia di sicurezza) e sulle interazioni con altri medicinali.
Conseguenza della ridefinizione: a tutt’oggi non è stato scientificamente provato se i “vaccini”-Covid-19 a base di mRNA (tra cui COMIRNATY di BioNTech) somministrati in massa non siano poi genotossici o cancerogeni. Questo perché sono stati omessi studi fondamentali”.[1] E finora non è stato escluso il loro effetto mutageno (cioè il danno permanente al DNA). Al contrario, vedi sotto.
COMIRNATY è una sostanza sperimentale a base di mRNA che non ha assolutamente nulla a che vedere con i vaccini convenzionali in termini di modalità d’azione e produzione.
La decisione di esecuzione della Commissione europea del 21 dicembre 2020 (doc. A. 5), che ha inizialmente autorizzato in via condizionata Comirnaty (“vaccino Covid-19 a mRNA (modificato a livello dei nucleosidi)” per l’uso generale, anche di massa, recita testualmente
“4) Il comitato per i medicinali per uso umano ha concluso che “RNA messaggero a singola elica con capping in 5’, prodotto mediante trascrizione in vitro senza l’ausilio di cellule (cell-free) dai corrispondenti DNA stampo, che codifica per la proteina virale spike (S) del SARS-CoV-2″ è una nuova sostanza attiva.”
Nell’allegato I, punto 5 (proprietà farmacologiche) della decisione di esecuzione del 10.10.2022 qui contestata (doc. A.2), il meccanismo d’azione è indicato come segue:
“L’RNA messaggero modificato a livello dei nucleosidi presente in Comirnaty è formulato in nanoparticelle lipidiche, per consentire il rilascio dell’RNA non replicante all’interno delle cellule ospiti e dirigere l’espressione transitoria dell’antigene S di SARS-CoV-2. L’mRNA codifica per una proteina S intera ancorata alla membrana …”
I nucleosidi sono i materiali di costruzione dell’RNA. L’RNA è un acido nucleico ed è essenziale per la sintesi delle proteine. Il progetto delle proteine nel corpo umano è memorizzato nel genoma umano, nel DNA nel nucleo della cellula, dove viene trascritto in mRNA. Una volta formato l’mRNA con il progetto di costruzione della proteina, l’mRNA lascia il nucleo della cellula. Al di fuori del nucleo cellulare, i ribosomi leggono questo piano di costruzione e formano la proteina corrispondente. In una cellula umana ci sono più di centomila molecole di mRNA contemporaneamente. I ribosomi riescono a leggere le informazioni solo in breve tempo perché l’mRNA di solito decade rapidamente.
Nel caso del “vaccino a mRNA”, invece, l’mRNA viene prodotto sinteticamente in laboratorio. Secondo l’osservazione effettuata ormai da due anni – che è in netta contraddizione con l’affermazione fatta ufficialmente alla popolazione che questo mRNA sintetico sarebbe rimasto nel muscolo superiore del braccio (dove avviene l’iniezione) (affermazione falsa, che è anche nei documenti ufficiali di approvazione, vedi sopra) – questa sostanza entra in tutto il corpo, e può persino attraversare la barriera emato-encefalica (Nature Neuroscience, The S1 protein of SARS-CoV-2 crosses the blood-brain barrier in mice, Elizabeth M. Rhea et al) ed è stata trovata nel corpo di persone trattate con questa sostanza anche mesi dopo l’iniezione. Dopo che alcune particelle sono state assorbite e la proteina spike è stata prodotta da esse, questa proteina spike può facilitare il passaggio di ulteriori particelle “vaccinali” nel cervello (A Case Report: Multifocal Necrotizing Encephalitis and Myocardities after BNT162b2 mRNA Vaccination against Covid-19).
Non solo c’è grande preoccupazione, ma anche prove evidenti che l’mRNA sintetico iniettato nel corpo può retrotrascriversi in DNA e che queste copie di DNA possono inserirsi nel DNA delle cellule umane, cioè nel genoma umano. Pertanto, l’informazione genetica dell’RNA può contaminare e alterare il genoma umano (Intercellular Reverse Transcription of Pfizer BioNTech COVID-19 mRNA Vaccine BNT162b in vitro in human liver cell line, Markus Alden et al; Potential mechanisms for human genome integration of Genetic Code from SARS-CoV-2 mRNA vaccination: implication for disease – Kyriakopoulos et. al).
La relazione scientifica “The immunological and biochemical principles of mRNA vaccine toxicity” di due microbiologi e uno specialista polmonare (Dr.med. Michael Palmer, Prof.Dr.med. Sucharit Bhakdi, Dr.med. Wolfgang Wodarg – Doc. A. 14) dimostra che la trascrizione inversa dell’RNA in DNA è un meccanismo noto da molti decenni (dagli anni ’70 del secolo scorso)! Pertanto, nulla di nuovo e soprattutto nulla che possa essere semplicemente escluso. Al contrario! Il rischio di trascrizione inversa aumenta, ovviamente, con ogni ulteriore iniezione.
Gli esperti scrivono: „Apparently, EMA’s experts were assuming that RNA in general will not affect the integrity of the host cell genome. The first exception to this rule has been known since 1970 … it could hardly be considered a novelty in 2020”.[2]
La sostanza a base di mRNA COMIRNATY è stata “formalmente classificata” come “vaccino”, anche se, come dimostrano i fatti, non svolge in alcun modo la funzione di un vaccino. Ovviamente, la sostanza a base di mRNA COMIRNATY è una sostanza che è stata classificata come “vaccino” sulla base di una “etichettatura di convenienza”, anche se non ha la funzione di una vaccinazione convenzionale, ma è un pro-farmaco che è costruito e agisce come un farmaco di terapia genica e deve quindi essere classificato tra i medicinali di terapia avanzata.
Sebbene COMIRNATY sia stato formalmente definito dall’EMA come un vaccino contro una malattia infettiva, e quindi non si qualificherebbe come un medicinale di terapia genica ai sensi della Direttiva 2009/120/CE della Commissione del 14 settembre 2009 e della Direttiva 2001/83/CE Allegato IV, punto 2.1. ultima frase, l’effettiva natura e modalità d’azione di COMIRNATY è quella di un medicinale di terapia genica. È quindi necessario fare riferimento alle disposizioni previste dal legislatore europeo per questa particolare categoria di prodotti.
La principale differenza tra la procedura di autorizzazione all’immissione in commercio dei medicinali per terapie avanzate (compresi i medicinali per terapia genica) e quella dei vaccini convenzionali può essere riassunta come segue.
Per i medicinali di terapia genica, l’allegato I, parte IV, della direttiva 2001/83 prevede, tra l’altro, quanto segue:
- Introduzione: …. I fattori di rischio da considerare comprendono: …. il livello di integrazione delle sequenze di acidi nucleici o dei geni nel genoma, la funzionalità a lungo termine, il rischio di oncogenicità e le modalità di somministrazione o uso ….
Requisiti speciali per il modulo 3
- Requisiti speciali
Oltre ai requisiti di cui ai punti 3.2.1. e 3.3.3. della parte I del presente allegato, si applicano i seguenti requisiti:
- Devono essere fornite informazioni su tutte le materie prime utilizzate per la fabbricazione della sostanza attiva, compresi i prodotti necessari per la modificazione genetica delle cellule umane …….
” 4.2 Requisiti speciali per i medicinali di terapia genica
4.2.1 Farmacologia
- Devono essere forniti studi in vitro e in vivo degli effetti relativi allo scopo terapeutico proposto (ossia studi farmacodinamici di prova) utilizzando modelli dedicati e specie di animali pertinenti che dimostrino che la sequenza di acido nucleico raggiunge il bersaglio previsto (organo o cellule bersaglio) e svolge la funzione prevista (livello di espressione e attività funzionale). Devono essere indicati la durata della funzione della sequenza di acido nucleico e il regime di dosaggio proposto negli studi clinici.
- Selettività del bersaglio: se un medicinale di terapia genica è destinato a svolgere una funzione selettiva o limitata al bersaglio, devono essere forniti studi che confermino la specificità e la durata della funzione e dell’attività nelle cellule e nei tessuti bersaglio.
Nota: contrariamente a quanto affermato al pubblico che la sostanza iniettata sarebbe rimasta nel muscolo superiore del braccio interessato e che la formazione della proteina spike si sarebbe concentrata lì, sia i nanolipidi che la proteina spike sono stati rilevati in tutto il corpo umano! Palmer et al. nella loro perizia sui vaccini a mRNA in generale (Doc. A.14) commentano:
„2.1. mRNA vaccines are distributed throughout the body and prominently affect the blood vessels”. The assertion that the mRNA/lipid nanoparticles remain at the site of injection is now widely known to be a blatant untruth. The “vaccines” rapidly spread from the site of injection to regional lymph nodes and to the blood circulation … Moreover, in contrast to most viruses, mRNA vaccine nanoparticles can be taken up by any cell type, including the endothelia, which form of the innermost cell layer of the blood vessels…. 2.2. The expression of spike protein in the body is widespread and long-lasting. Studies on a model mRNA vaccine have shown that the lipid nanoparticles, after intramuscular injection, rapidly enter the bloodstream. They subsequently accumulate preferentially in certain organs including the liver, the spleen, and the ovaries. … at least the blood vessels themselves are exposed to the vaccine in every organ and every tissue, from which we have to expect widespread expression of the foreign antigen… Another important consideration is how soon the antigen is expressed, and how long this expression lasts….a fairly long-lasting expression of spike after mRNA vaccination was also reported by Röltgen et al., wo still detected the spike protein in lympfh nodes 60 days after the second injection, and at this same time point also showed the continued presence of mRNA encoding the spike. Similarly, Magen et al. detected strong spike protein expression and continued presence of the RNA at one month after vaccination….”[3]
4.2.2 Farmacocinetica
- a) Gli studi di biodistribuzione devono comprendere studi di persistenza, clearance e mobilizzazione. Gli studi di biodistribuzione devono anche considerare il rischio di trasferimento del gene nella linea germinale.
Nota: Palmer et al nella loro opinione di esperti sui vaccini a mRNA in generale (Doc. A.14): „4.2 Pharmacokinetiks of mRNA vaccines. The properties of the lipid nanoparticles … exert a strong influence on their transport and their fate within the human body. 4.2.1 Organ distribution of model mRNA vaccines. … the transport of vaccine lipid nanoparticles may resemble that of lipoproteins … the amount of lipoprotein particles taken up und turned over varies greatly between the cells of different organs. The following organs take up particularly large amounts:
- The liver has a central place in lipoprotein metabolism…
- Endocrine glands which produce steroid hormones … These includes the testes, the ovaries, and the adrenal glands,
- The placenta requires lipoprotein both for supplying the fetus and for its won production of progestin hormones, which are necessary to sustain pregnancy,
- The lacting breast glands acquire fat und cholesterol from lipoproteins and repackage them for release into the breast milk.
With this in mind, we can understand some of the observations on the distribution of mRNA vaccines within the body …Moderna, according to EMA’s report on this vaccine, … submitted some animal data on a model vaccine … In this study, the levels o mRNA rather than of the lipids were measured. The results of Moderna’s study are incompletely described in the report, but on page 47 we read:
Increased mRNA concentrations (compared to plasma levels) were found in the spleen and eye. … Low levels of mRNA could be detected in all examined tissues except the kidney. This included heart, lung, testis and also brain tissues … liver distribution of mRNA-1647 is also evident in this study, consistent with the literature reports that liver is a common target organ of LNPs.” …
regardless of the tissue in any specific organ, at least the blood vessels and their endothelia will be exposed to the vaccine particles in each and every organ. Accordingly, vasculitis and thromboembolic events are somewhat likely to occur in all organs. Additional tissue-specific pathology might be expected to focus on organs with high levels of accumulation. However, as we will see presently, the findings of these animal studies likely do not give a complete picture of mRNA vaccine distribution in practice. 4.2.2. Correlation of model vaccine organ distribution with histopathological findings … we have seen evidence of inflammation and of vaccine-induced spike protein expression in heart muscle .. and the brain …, even though these organs accumulated only comparatively low or moderate levels of the model vaccine in Pfizer’s and Moderna’s animal experiments. The observed inflammation is particularly remarkable with respect to the brain, which is supposed to be protected by the blood-brain barrier. In this context, we must note two important caveats: 1. The blood-brain barrier breaks down when the brain tissue is afflicted by inflammation. Accordingly, vasculitis within the brain that was induced by the first injection of an mRNA vaccine might soften up the blood-brain barrier and facilitate the entry of vaccine particles delivered with a subsequent booster injection. It would therefore have been important to examine the organ distribution of the vaccine not only after the first injection, but also after one or more repeat injections. However, this was not done in Pfizer’s and Moderna’s animal studies.
- The SARS-CoV-2 spike protein has been shown in several studies to compromise the integrity of the blood-brain-barrier … Spike protein which may be expressed elsewhere but reaches the brain through the bloodstream may facilitate penetration of vaccine particles into the brain…. These considerations, in combination with histopathological findings, strongly suggest that mRNA vaccines distribute more widely and effectively than Pfizer’s and Moderna’s very limited animal studies on model vaccine would indicate…
4.2.3. Time course of elimination and duration of activity. We had seen in Section 4.1.4. that the mRNA can become separated from the lipids after the cellular uptake of the vaccine nanoparticles. The elimination of both ingredients must therefore be considered separately.
4.2.3.1. Time course of mRNA elimination. … it must be stressed out that none of these studies used the mRNA deployed in the COVID-19 vaccines, and furthermore that all studies were carried out in rodents. These results can therefore not be directly applied to the current crop of mRNA vaccines and their use in human patients. … Covid-19 vaccine mRNA has been detected at 60 days after injection in lymph nodes … and at 30 day within muscle tissue of a limb other than the one which had been injected … Long-lasting persistence of the vaccine mRNA in blood plasma samples of injected patients was recently reported by Fertig et al. … these studies on humans show that the vaccine mRNAs may persist much longer than Pfizer’s and Moderna’s animal studies would suggest.
4.2.3.2. Time course of lipid elimination. …According to EMA report .. Moderna submitted no data on the elimination of the two synthetic lipids contained in their Covid-19 mRNA vaccine. … While EMA reassures us that accumulation of the lipids within the body is unlikely, we must note that firstly the information provided is entirely insufficient by the usual standards of drug development and approval, and secondly that absence of lipid accumulation does not imply absence of cumulative toxicity.”[4]
(b) Nel contesto delle valutazioni del rischio ambientale, devono essere forniti studi sull’escrezione e sul rischio di trasmissione a terzi, altrimenti ciò deve essere debitamente giustificato nella domanda sulla base della natura del medicinale in questione.
4.2.3. Tossicologia
(a) Deve essere valutata la tossicità del medicinale di terapia genica finito. Inoltre, a seconda del tipo di medicinale, le sostanze attive e gli eccipienti devono essere testati separatamente e deve essere valutato l’effetto in vivo di prodotti non destinati alla funzione fisiologica ma codificati dalla sequenza di acido nucleico.
Nota: Palmer et al nella loro revisione dei vaccini a mRNA in generale (Doc. A.14):
“): 4.3. Lipid nanoparticle toxicity. … two synthetic lipid species. The PEG-conjugated lipids are the less abundant of the two, and the only mechanism of harm on record consists in allergic reactions to these lipids. In contrast, the cationic lipids account for almost half of the total lipid in the vaccine LNPs, and they can exert toxicity outright, without any “assistance” from the adaptive immune system. …
4.3.2. Inflammatory signaling by cationic lipids. Several experimental studies have shown that cationic lipids similar to those used in the Pfizer and Moderna COVID-19 vaccines induce strong inflammatory reactions. … This agrees with the frequent observation of local and also systemic inflammatory reactions among COVID-19 vaccine recipients….”
- Genotoxicity of mRNA vaccines … 5.2.1.4. Summary. Even though this had not yet been experimentally demonstrated when the COVID-19 mRNA vaccines were given emergency approval, there was ample precedent to suggest the strong possibility that DNA copies of the vaccine mRNA would arise and be inserted into the cellular genome. Rather than waving away this risk as they did, EMA and other regulators should have obligated Pifzer and Moderna to carry out the necessary studies for excluding this risk before green-lighting authorization…The results reported by Aldén et al., even though preliminary in some respects, pose some very serious questions that can no longer be ignored by the regulatory authorities…. Gene inactivation. Insertion may occur within a gene and disrupt it. This can lead to the loss of important cellular gene products (i.e., proteins) and thus, potentially, to the development of disease including cancer. … Gene regulation. Transcriptional and epigenetic regulation mechanisms may be affected, thus modulating protein expression levels upward and downward with unpredictable and undesirable results. Indirect regulatory effects may effect even distant genes located on other chromosomes.”… Activation of oncogenes… the occurrence of malignancies through DNA integration and activation of cancer-promoting genes (oncogenes) has been demonstrated in clinical trials … for the genetic treatment of children …. These malignancies will typically become manifest only several years after the completion of treatment. Therefore, thorough long-term investigations concerning possible genotoxic effects of the chromosomal integration are absolutely necessary, in both the pre-clinical and the clinical trial stages, for a valid benefit-risk analysis…. The risk of insertion into the chromosomal DNA must be taken seriously…. Autoimmune-like disease. Integration of the spike protein gene into the host cell could lead to permanent expression of this antigen and thus induce chronic autoimmune-like disease… Germline integration. … Pfizer’s own experiments indicate a high level of vaccine accumulation in the ovaries … Furthermore, LINE-1 and other retrotransposons are active and cause genomic insertion events in human oocytes … In combination, these findings, indicate that the mRNA gene sequences may be integrated into the DNA of oocytes, and hence into the human germline. Insertion into male germline cells cannot be ruled out either, even though in the cited animal study the tissue levels of the model mRNA vaccine in the testes was significantly lower than in the ovaries. Should this indeed come to pass – should the germline cells of vaccinated individuals be rendered transgenic – then the risk of spawning or conceiving transgenic children will not be limited to these individuals only, but it will necessarily be shared by their current or future spouses. In effect, an entire generation of future parents will be exposed to this risk. … Summary. Integration of the mRNA sequences into somatic cells is likely and implies a risk of cancer and of autoimmune disease. Moreover, the risk of germline integration, resulting in transgenic offspring, cannot be denied. These risks must urgently be addressed through in depth-animal studies. Meanwhile, the authorizations of any and all mRNA vaccines in current use must urgently be revoked.”[5]
(b) gli studi di tossicità della somministrazione a dose singola possono essere combinati con studi di sicurezza farmacologica e farmacocinetica, ad esempio sulla persistenza.
(c) Devono essere forniti studi di tossicità della somministrazione a dose ripetuta se si prevede una somministrazione multiplo all’uomo. La via e il programma di somministrazione devono essere strettamente allineati al dosaggio clinico previsto. Nei casi in cui una singola dose può determinare una funzione sostenuta della sequenza di acidi nucleici nell’uomo, devono essere presi in considerazione studi di tossicità a dose ripetuta. Questi studi possono essere più lunghi degli studi di tossicità standard, a seconda della persistenza del medicinale di terapia genica e dei rischi potenziali previsti. La durata deve essere giustificata.
- d) Deve essere studiata la genotossicità. …[6]
- e) La cancerogenicità deve essere studiata[7] . … a seconda del tipo di medicinale, … il potenziale tumorale deve essere valutato in modelli pertinenti in vivo/in vitro.
Nota: si veda la nota sopra riportata alla voce Tossicità e inoltre Prof. S. Bhakdi et al. nel loro specifico parere scientifico su Comirnaty e il suo uso “Expert statement regarding Comirnaty – COVID-19 mRNA vaccine for children” (Doc. A.15):
- “2.3. Genotoxicity
No studies have been carried out regarding genotoxicity, that is, damage to the human genetic material, which could lead to heritable mutations and cancer. In the EMA report …, this is justified as follows:
No genotoxicity studies have been provided. This is acceptable because the components of the vaccine formulation are lipids and RNA, which are not expected to have genotoxic potential. The risk assessment performed by the applicant shows that the risk of genotoxicity related to these excipients [i.e. the synthetic lipids] is very low based on literature data.
In reality, it is known that the LNPs contained in BNT162b2 can enter all kinds of cells—that is, after all, the purpose of their inclusion in this vaccine preparation. It is also known that, once inside the cell, cationic lipids disrupt mitochondrial function (cell respiration) and cause oxidative stress, which in turn leads to DNA damage.
It should be mentioned that two of the lipids used by Pfizer—namely, the cationic lipid ALC-0315 and the PEGylated lipid ALC-0159, which account for 30-50% and for 2-6%, respectively, of the total lipid content—had not previously been approved for use in humans. Pfizer’s and EMA’s cavalier attitude to the use of novel and so far unproven chemicals as components in drug or vaccine preparations without comprehensive studies on toxicity, including genotoxcicity, is completely unscientific and unacceptable.”[8]
(f) Tossicità per la riproduzione e lo sviluppo: devono essere forniti studi sugli effetti sulla fertilità e sulla funzione riproduttiva generale. Devono essere forniti anche studi sulla tossicità embrionale, fetale e perinatale e studi sulla trasmissione germinale; …
Nota: su questo punto il Prof. S. Bhakdi et al. nella loro relazione di esperti preparata appositamente per la COMIRNATY (Doc. A.15):
- “1.1.7. Potential risks to fertility and to the breastfed newborn
A high level of expression of spike in the ovaries raises the prospect of significant damage to that organ, with possible consequences for female fertility. Uptake of the vaccine by mammary gland cells opens two possible pathways of toxicity to the breastfed child: firstly, the expression of spike protein and its secretion into the breast milk, and secondly, the wholesale transfer of the vaccine into the milk. The mammary glands are apocrine, which means that they pinch off and release fragments of their own cytoplasm into the milk; thus, anything that has reached the cytoplasm might also reach the breast milk. In this connection, we note that both the VAERS database and the EU drug adverse events registry (EudraVigilance) report fatalities in breastfed newborns after vaccination of their mothers (see Section 3.1.3.6). ….
- 2.4. Reproductive toxicity
Reproductive toxicity was assessed using only one species (rats) and on only small numbers of animals (21 litters). A greater than twofold increase in pre-implantation loss of embryos was noted, with a rate of 9.77% in the vaccine group, compared to 4.09% in the control group. Instead of merely stating …that the higher value was “within historical control data range,” the study should have stated unambiguously whether or not this difference was statistically significant; and if it was not, the number of experiments should have been increased to ensure the required statistical power. The same applies to the observations of “very low incidence of gastroschisis, mouth/jaw malformations, right sided aortic arch, and cervical vertebrae abnormalities.” Overall, these studies are inadequately described and apparently were also inadequately carried out. “[9]
(g) Studi supplementari sulla tossicità
– Studi di integrazione: gli studi di integrazione devono essere forniti per ogni medicinale di terapia genica, a meno che la loro assenza non sia scientificamente giustificata, ad esempio perché le sequenze di acido nucleico non entrano nel nucleo. Per i medicinali di terapia genica che non si prevede siano in grado di integrarsi, gli studi di integrazione devono comunque essere eseguiti se i dati di biodistribuzione indicano un rischio di trasmissione germinale.
Nota: vedere anche la nota al punto 4.2.2. Farmacocinetica a)
– Immonogenicità e immunotossicità: devono essere studiati i potenziali effetti immunogenici e immunotossici. …
- requisiti speciali per il modulo 5
5.1 Requisiti specifici per tutti i medicinali per terapie avanzate
5.1.1. i requisiti specifici della presente sezione della parte IV si aggiungono ai requisiti stabiliti nel modulo 5 della parte 1 del presente allegato….
5.1.6 L’efficacia deve essere dimostrata, in relazione all’uso previsto, dai risultati pertinenti di studi clinici che utilizzano endpoint clinicamente significativi per l’uso previsto. In alcune circostanze cliniche può essere richiesta la prova dell’efficacia a lungo termine. Deve essere presentata la strategia per la valutazione dell’efficacia a lungo termine.
5.1.7 Nel piano di gestione del rischio deve essere inclusa una strategia per il monitoraggio a lungo termine della sicurezza e dell’efficacia.
5.2 Requisiti speciali per i medicinali di terapia genica
5.2.1 Studi di farmacocinetica nell’uomo
Gli studi di fiammacocinetica sull’uomo devono includere quanto segue:
- a) studi di disseminazione per valutare l’escrezione dei medicinali di terapia genica;
- b) studi di biodistribuzione;
- c) studi farmacocinetici del medicinale e delle frazioni di espressione genica (ad esempio proteine espresse o firme genomiche).
5.2.2. Studi di farmacodinamica nell’uomo
Gli studi di farmacodinamica nell’uomo valutano l’espressione e la funzione della sequenza di acido nucleico successivamente alla somministrazione del medicinale di terapia genica.
5.2.3 Studi sulla sicurezza
Negli studi sulla sicurezza si deve indagare su quanto segue: …
- c) la ricombinazione delle sequenze genomiche esistenti;
- d) la proliferazione neoplastica dovuta a mutagenicità inserzionale.
Per quanto riguarda i “vaccini”, il Codice Comunitario dei Medicinali per Uso Umano (Direttiva 2001/83/CE) prevede solo le seguenti norme molto scarne, che peraltro si riferiscono tutte esclusivamente a vaccini convenzionali basati su antigeni e non hanno nulla in comune con le iniezioni di mRNA come COMIRNATY.
I vaccini sono classificati come medicinali biologici nella parte III dell’allegato I della direttiva 2011/83/CE.
Al punto 1.2, il Codice europeo dei medicinali definisce i requisiti per l’autorizzazione dei vaccini nella Parte III del suo Allegato, ma si riferisce esclusivamente a sostanze basate su un antigene.
Non si fa assolutamente menzione dei requisiti aggiuntivi (oltre a quelli generali) per i prodotti terapeutici avanzati nel caso dei vaccini!
L’affermazione giuridica secondo cui, a prescindere dalla loro composizione effettiva e dalla loro modalità d’azione, i “vaccini contro le malattie infettive” non sarebbero terapie genetiche deve essere riconosciuta e accertata come scientificamente infondata e i passaggi corrispondenti della Direttiva 2001/83/CE del Parlamento europeo e del Consiglio (cioè l’Allegato I, Parte IV, punto 2.1, ultima frase) e della Direttiva 2009/20/CE della Commissione (cioè l’Allegato IV, punto 2.1, ultimo paragrafo) devono essere accertati e dichiarati come gravemente illegali ai sensi del diritto dell’UE, con le relative necessarie conseguenze.
Inoltre, sulla base di quanto sopra, deve essere accertata e dichiarata la grave illegittimità della procedura di autorizzazione (sia quella per l’autorizzazione condizionata che quella per l’autorizzazione non più condizionata) nonchè dell’autorizzazione all’immissione in commercio (la prima condizionata e l’attuale non più condizionata) e, di conseguenza, devono essere dichiarate nulle le decisioni di esecuzione della Commissione qui impugnate. Inoltre, le direttive impugnate devono essere annullate nella parte pertinente per violazione dei principi imperativi del diritto farmaceutico.
(2)
Va inoltre ricordato che il legislatore dell’UE stabilisce in ogni caso che il Comitato per le terapie avanzate deve essere coinvolto nella procedura di autorizzazione all’immissione in commercio di medicinali che, anche se non dovessero essere classificati come medicinali per terapie avanzate, funzionano comunque per aspetti essenziali come questi (come è il caso di COMIRNATY)!
Nei considerando (8), (10), (11) (12) (13) (20) del Regolamento (CE) n. 1394/2007 del Parlamento europeo e del Consiglio, del 13 novembre 2007, sui medicinali per terapie avanzate, il legislatore dell’UE prevede quanto segue:
(8) Il presente regolamento rispetta i diritti fondamentali e osserva i principi figuranti nella Carta dei diritti fondamentali dell’Unione europea e tiene conto della Convenzione del Consiglio d’Europa per la protezione dei diritti dell’uomo e della dignità dell’essere umano riguardo all’applicazione della biologia e della medicina: Convenzione sui diritti umani e la biomedicina.
(10) “La valutazione dei medicinali per terapie avanzate richiede spesso competenze molto specifiche che vanno oltre il campo della farmacologia tradizionale e toccano altre aree di competenza come la biotecnologia o i dispositivi medici. È quindi opportuno istituire un comitato per le terapie avanzate all’interno dell’Agenzia, che dovrebbe essere incaricato di preparare un progetto di parere sulla qualità, la sicurezza e l’efficacia del medicinale per terapia avanzata in questione e di presentarlo al comitato per i medicinali per uso umano per l’approvazione. Inoltre, il Comitato per le terapie avanzate dovrebbe essere consultato in relazione alla valutazione di altri medicinali quando è richiesta una specifica esperienza nel suo campo di competenza“.
(11) “Il comitato per le terapie avanzate deve riunire le migliori competenze disponibili nella Comunità in materia di medicinali per terapie avanzate. La composizione del comitato per le terapie avanzate deve garantire un’adeguata copertura delle aree di competenza scientifica relative alle terapie avanzate, tra cui la terapia genica …., la farmacovigilanza e l’etica. Dovrebbero essere rappresentate anche le organizzazioni dei pazienti e i clinici con competenze scientifiche sui medicinali per terapie avanzate”.
(12) “Per garantire la coerenza scientifica e l’efficienza del sistema, l’Agenzia deve assicurare il coordinamento tra il comitato per le terapie avanzate e gli altri comitati, gruppi consultivi e gruppi di lavoro, in particolare il comitato per i medicinali per uso umano…”.
(13) “I medicinali per terapie avanzate devono essere soggetti agli stessi principi normativi degli altri tipi di medicinali biotecnologici. Tuttavia, i requisiti tecnici, in particolare la natura e la portata dei dati preclinici e clinici relativi alla qualità per dimostrare la qualità, la sicurezza e l’efficacia del medicinale, possono essere altamente specifici. Per i medicinali di terapia genica … questi requisiti sono già stabiliti nell’Allegato I della Direttiva 2001/83/CE …
(20) Il monitoraggio dell’efficacia e degli effetti collaterali è un aspetto cruciale per la regolamentazione dei medicinali per terapie avanzate. Il richiedente deve quindi descrivere dettagliatamente nella domanda di autorizzazione all’immissione in commercio se sono previste misure per garantire tale controllo e quali sono tali misure. Se giustificato da motivi di salute pubblica, il titolare dell’autorizzazione all’immissione in commercio dovrebbe anche essere tenuto a mettere in atto un adeguato sistema di gestione del rischio per affrontare i rischi associati ai medicinali per terapie avanzate.”
A causa della definizione abusiva di “vaccino”, come descritto sopra al punto (1), o dell’esclusione puramente formale di tutte le sostanze formalmente definite come “vaccini contro le malattie infettive” – che non corrisponde alle circostanze di fatto – anche in contrasto con le proprietà e gli effetti di fatto, COMIRNATY non è stato sottoposto a una serie di studi essenziali da parte di BioNTech, che sono invece indispensabili al fine di determinare l’efficacia e la sicurezza di una sostanza che agisce di fatto come un agente di terapia genica!
Anche se si volesse ipotizzare che COMIRNATY non dovrebbe essere classificato come farmaco per la terapia genica, a causa della sua composizione (sostanza di acido nucleico impacchettata in particelle nano-lipidiche) e della sua modalità d’azione (mediante iniezione della sostanza di acido nucleico, induzione, nell’ambito dell’espressione genica, della produzione della proteina spike e quindi di una tossina), il Comitato per le terapie avanzate avrebbe comunque dovuto essere coinvolto nella procedura di autorizzazione, poiché, indiscutibilmente, COMIRNATY ha proprietà e modalità d’azione che possono essere adeguatamente valutate solo da questo comitato speciale!
Il Prof. S. Bhakdi et al. hanno spiegato quanto segue nella loro dichiarazione di esperti (Doc. A.15):
“3.1.1. …
Comirnaty, like all other gene-based COVID-19 vaccines, causes the expression in vivo of one structural protein of SARS-CoV-2 – namely, the so-called spike protein, which naturally occurs on the surface of the virus particle. ….The key idea behind the Comirnaty vaccine is as follows:
-
- a synthetic mRNA that encodes the spike protein is complexed with a mixture of neutral and cationic (positively charged) synthetic lipids, which cluster together in lipid nanoparticles (LNPs);
- after injection, the LNPs facilitate the uptake of the mRNA into host cells, where the mRNA will cause the expression (synthesis) of the spike protein;
- the spike protein will appear on the surface of the host cells and induce an immune reaction itself.”[10]
Il Comitato per le terapie avanzate avrebbe dovuto essere coinvolto nella procedura di approvazione di COMIRNATY in ogni caso, a causa della composizione e del modo d’azione della sostanza, indipendentemente dalla definizione legale di “vaccino” contro una malattia infettiva, in conformità con la necessità stabilita dal legislatore dell’UE nel considerando (10) del Regolamento (CE) n. 1394/2007! Ma questo non è accaduto!
Il 22 luglio 2022, l’avvocato Renate Holzeisen, anche in nome e per conto di Children’s Health Defense Europe, ha presentato alla Commissione UE e all’EMA una istanza di ostensione (esibizione) degli studi condotti dall’EMA, per le sostanze Spikevax di Moderna e Comirnaty di Pfizer/BioNTech, sulla genotossicità, cancerogenicità e mutagenicità, nonché sul coinvolgimento nel processo di approvazione del Comitato per le terapie avanzate (Doc. A. 16).
In data 21.9.2022, l’EMA ha fornito la prima parte della risposta (A. 17) alla suddetta richiesta di divulgazione, affermando esplicitamente: „… please note that none of the authorised COVID-19 vaccines, including Comirnaty and Spikevax, have received input by EMA-S committee for advanced therapies (CAT) and therefore, no documentation proving the involvement of the CAT Committee in the respective procedure of the conditional marketing authorization (CMA) exist.
„Please note that mRNA COVID-19 vaccines are not gene therapy. According to the definition of gene therapy medicinal products in Annex I to Directive 2001/83/EC, PART IV, point 2.1., only such products are gene therapy medicinal products that are used in or administered to human beings with a view to regulating, repairing, replacing, adding or deleting a genetic sequence. None of this is the case for mRNA vaccines. Therefore, the involvement of the CAT was not foreseen for any COVID-19 vaccine”.[11]
L’EMA sostiene in modo lapidario che COMIRNATY (e Spikevax) non sarebbero prodotti per la terapia genica, ignorando completamente il fatto che i vaccini a base di mRNA non solo rientrano nella definizione di prodotti per la terapia genica a causa della loro composizione e modalità d’azione (come spiegato al punto (1)), ma che, indipendentemente dalla loro qualifica di medicinali per terapia genica, il coinvolgimento del comitato specifico per le terapie avanzate sarebbe stato obbligatorio nella procedura di autorizzazione per COMIRNATY (e Spikevax) a causa delle loro proprietà! Si veda il considerando (10) del Regolamento (CE) n. 1394/2007.
La volontà del legislatore è chiara a questo proposito! In ogni caso, il Comitato per le terapie avanzate avrebbe dovuto essere coinvolto nel procedimento di autorizzazione!
Ma questo è esattamente ciò che non è accaduto! Anche solo per questo motivo, il procedimento di autorizzazione è gravemente illegale e deve essere dichiarata la nullità delle decisioni della Commissione UE qui contestate.
(3)
Persino i requisiti più severi per i vaccini basati sull’ingegneria genetica rispetto a quelli per i vaccini convenzionali sono stati grossolanamente violati dall’EMA e dalla Commissione!
L’incredibile audacia nel brutale disprezzo dei principi fondamentali del diritto farmaceutico arriva persino al punto che le sostanze a base di mRNA come COMIRNATY e Spikevax sono state e sono deliberatamente trattate dall’EMA e dalla Commissione Europea come vaccini convenzionali nella procedura di autorizzazione, senza che siano stati applicati nemmeno i requisiti più severi per i vaccini basati sull’ingegneria genetica!
Per quanto riguarda gli studi di genotossicità, l’EMA risponde (doc. A. 17) come segue: “…Please note that no genotoxicity nor carcinogenicity studies have been submitted by the applicant of Comirnaty, as the components of the vaccine formulation are lipids and RNA that are not expected to have genotoxic potential. ….Genotoxicity or carcinogenicity studies are usually not required for the final vaccine formulation and therefore are not normally requested from the applicant. This is in line with the two WHO guidelines on the nonclinical evaluation of vaccines (WHO 2005, WHO 2013).”[12]
L’EMA e la Commissione Europea hanno quindi dichiarato esplicitamente di aver violato la linea guida specifica per i vaccini a DNA, perché si riferiscono esplicitamente ed esclusivamente alla linea guida dell’OMS per i vaccini convenzionali, che però non hanno assolutamente nulla a che fare con i vaccini a mRNA.
Va inoltre sottolineato che le linee guida dell’OMS in linea di principio non sono vincolanti in alcun modo, e quindi il semplice riferimento alle linee guida dell’OMS per i vaccini convenzionali è ancora più assolutamente inaccettabile!
Inoltre, l’EMA afferma in modo lapidario che non sono stati effettuati studi di genotossicità e cancerogenicità perché non si prevedeva che i componenti della sostanza (lipidi e RNA) avessero un potenziale genotossico! Con questa affermazione, l’EMA conferma che “si sta giocando alla roulette russa” con l’intera ignara popolazione dell’UE (e i suoi discendenti).
La regolamentazione legale dei vaccini non convenzionali (basati sull’ingegneria genetica) richiede studi molto più approfonditi di quelli previsti per i vaccini convenzionali.
“Secondo il rapporto di valutazione (Assessment Report) dell’EMA, gli studi mancanti:
I seguenti studi preclinici, che secondo lo stato delle conoscenze scientifiche avrebbero dovuto essere condotti prima della sperimentazione clinica, non sono mai stati effettuati per COMIRNATY:
- sulla farmacodinamica secondaria,
- sulla farmacologia della sicurezza con la “Core Battery” di studi sugli effetti sulla salute dei pazienti
- cardiovascolare
- sistema nervoso e
- sistema respiratorio.
- sulla farmacocinetica con
- assorbimento / biodisponibilità,
- distribuzione (“distribuzione in funzione del tempo della sostanza in esame in diversi organi e componenti corpuscolari del sangue, nonché legame con le proteine plasmatiche”) e
- metabolismo, compreso il tratto gastrointestinale e il primo passaggio epatico
in 2 diversi modelli animali (1 roditore e 1 non roditore).
- sulla tossicologia con
- genotossicità nella misura in cui deve essere possibile valutare la genotossicità della sostanza in esame (ad esempio, mediante studi di mutazione genica nei batteri);
- cancerogenicità nella misura in cui deve essere possibile valutare la cancerogenicità della sostanza in esame;
- tossicità per la riproduzione e lo sviluppo, nella misura in cui il dossier deve consentire la valutazione degli effetti della sostanza in esame sugli organi riproduttivi maschili.
- antigenicità e immunotossicità, se applicabile.
Giustificazione da parte dell’EMA dell’omissione degli studi sulla farmacologia di sicurezza e sulla genotossicità e cancerogenicità
Il rapporto di valutazione dell’EMA a pag. 45 (RAPPORTO DI VALUTAZIONE – Doc. A.25) afferma quanto segue:
„Safety pharmacology studies
No safety pharmacology studies were conducted with BNT162b2. The Applicant refers to that they are not considered necessary according to the WHO guideline (WHO, 2005). In addition, no findings on vital organ functions have been recorded in the repeat dose toxicology studies. Thus, the absence of safety pharmacology studies is endorsed by the CHMP.”[13]
Linea guida dell’OMS sulla valutazione non clinica dei vaccini (2005) e il suo predecessore europeo (CPMP/SWP/465/95).
La linea guida dell’OMS del 2005, WHO guideline on nonclinical evaluation of vaccines, WHO Technical Report Series, No. 927, 2005 (Doc. A.19) Anche nelle linee guida dell’UE, che incorporano in parte le linee guida ICH (linea guida sulla buona pratica clinica per l’UE, il Giappone e gli USA), non si fa riferimento alla linea guida dell’OMS del 2005.
Tuttavia, esisteva una nota dell’EMA (CPMP) per una guida sui test preclinici farmacologici e tossicologici dei vaccini del 1997 (CPMP/SWP/465/95 (Doc. A.22).
Questa linea guida contiene gli studi richiesti da un punto di vista scientifico per l’autorizzazione di nuovi vaccini. Il campo di applicazione è stato definito per i seguenti vaccini:
„Within the context of this Note for Guidance, new vaccines are those containing antigens not yet described in the European Pharmacopoeia monographs or in WHO requirements, or using a new conjugate for a known antigen, or any new combination of known and/or new antigens….. (S. 2) DNA-vaccines, gene therapy or genetically altered somatic cell therapy are not addressed in this Note for Guidance.” (S. 3)
Traduzione in Italiano:
“Nel contesto di questa Nota di orientamento, i nuovi vaccini sono quelli che contengono antigeni non ancora descritti nelle monografie della Farmacopea europea o nei requisiti dell’OMS, o che utilizzano un nuovo coniugato per un antigene noto, o qualsiasi nuova combinazione di antigeni noti e/o nuovi….. (p. 2) I vaccini a DNA, la terapia genica o la terapia con cellule somatiche geneticamente modificate non sono trattati in questa Nota di orientamento”. (p. 3)
La Nota di orientamento (Note for Guidance) escludeva esplicitamente dal suo campo di applicazione i vaccini a RNA, DNA e simili. Ciò era anche logico, dal momento che – come verrà spiegato di seguito – la specificità dei vaccini a base di DNA o geni era coperta dalla linea guida “CPMP note for guidance on the quality, preclinical and clinical aspects of gene transfer medicinal products (CPMP/BWP/3088/99) – Doc. A.20″.
Per i nuovi vaccini, questa linea guida considera necessari anche studi, ad esempio, sulla farmacodinamica secondaria, sulla farmacologia della sicurezza e sulla farmacocinetica:
- Farmacodinamica secondaria / farmacologia della sicurezza:
„The potential for undesirable pharmacological activities e.g. on the circulatory and respiratory systems should be considered for new vaccines (as defined in the Scope) and investigated in appropriate animal models.” (S. 5)
Traduzione:
“Il potenziale di attività farmacologiche indesiderate, ad esempio sul sistema circolatorio e respiratorio, dovrebbe essere preso in considerazione per i nuovi vaccini (come definito nel campo di applicazione) e studiato in modelli animali appropriati.” (p. 5)
- Farmacocinetica:
La necessità di studi farmacocinetici deve basarsi su una valutazione caso per caso. La novità della sostanza in questione gioca un ruolo decisivo, per cui per le nuove sostanze potrebbe essere necessario effettuare studi sulla biodistribuzione, sugli esami istopatologici dei linfonodi vicini al sito di iniezione e sull’escrezione virale. (S. 4)
Documento di domande e risposte sull’abrogazione della linea guida (CPMP/SWP/465)
Nella pagina dell’EMA (doc. A.21), questa guida è indicata come ritirata. In un documento di domande e risposte dell’EMA intitolato “Questions and answers on the withdrawal of the CPMP Note for guidance on preclinical pharmacological and toxicological testing of vaccines (CPMP/SWP/465)”, si spiega che la linea guida originale è stata ritirata e sostituita dalla linea guida dell’OMS del 2005:
„The WHO guideline on nonclinical evaluation of vaccines (2) was published in 2005 and was the result of a collaboration between experts from different regulatory agencies, health agencies, academic institutions and vaccine manufacturers. EU regulators took active part in this work. Following discussion within the CHMP Safety Working Party and Vaccine Working Party, it has been agreed to remove the CPMP guideline on preclinical pharmacological and toxicological testing of vaccines, and to refer to the WHO guideline on nonclinical evaluation of vaccines.”[14]
Per quanto riguarda la validità e il carattere, il documento Q & A afferma che le linee guida dell‘OMS, come quelle del CHMP, sono solo raccomandazioni e non sono vincolanti. Esse possono essere disattese con un’adeguata giustificazione:
„A CHMP guideline does not have legal force. As written in the EMA document describing the status of scientific guidelines: “Scientific guidelines are to be considered as a harmonised Community position, which if they are followed by relevant parties such as the applicants, marketing authorisation holders, sponsors, manufacturers and regulators will facilitate assessment, approval and control of medicinal products in the European Union. Nevertheless, alternative approaches may be taken, provided that these are appropriately justified.” (3). Therefore, by referring to the WHO guideline, the same principles apply.”[15]
Ambito di applicazione della linea guida OMS 2005
La linea guida dell’OMS definisce il suo campo di applicazione nella sezione 1.1 come segue:
„Vaccines for human use include one or more of the following: microorganisms inactivated by chemical and/or physical means that retain appropriate immunogenic properties; living microorganisms that have been selected for their attenuation whilst retaining immunogenic properties; antigens extracted from microorganisms, secreted by them or produced by recombinant DNA technology; chimeric microorganisms; antigens produced in vivo in the vaccinated host following administration of a live vector or nucleic acid or antigens produced by chemical synthesis in vitro. The antigens may be in their native state, truncated or modified following introduction of mutations, detoxified by chemical or physical means and/or aggregated, polymerized or conjugated to a carrier to increase immunogenicity. Antigens may be presented plain or in conjunction with an adjuvant, or in combination with other antigens, additives and other excipients.”[16]
La linea guida si concentra sugli “antigeni”. Gli “antigeni prodotti in vivo nell’ospite vaccinato in seguito alla somministrazione di un vettore vivo o di un acido nucleico” sono quegli antigeni che vengono prodotti dagli organismi dopo l’iniezione dell’mRNA al loro interno e poi ottenuti per estrazione. Un esempio è il vaccino Nuvaxovid, in cui le proteine di punta sono prodotte dalle larve di falena e poi “raccolte” o estratte per essere aggiunte al vaccino.
COMIRNATY NON è un antigene come definito in questa linea guida dell’OMS. Pertanto, questa linea guida NON si applica alle iniezioni di mRNA come COMIRNATY!
Tuttavia, anche se si dovesse sostenere l’applicabilità della linea guida, ulteriori indagini avrebbero dovuto essere condotte dal produttore Moderna prima dell’inizio della sperimentazione clinica.
Regolamento per i test farmacologici-tossicologici da effettuare
Dal punto 4.1 della linea guida (pag. 45) emerge che i seguenti parametri devono essere controllati nell’ambito degli studi tossicologici:
“Potential toxic effects of the product should be evaluated with regard to target organs, dose, route(s) of exposure, duration and frequency of exposure, and potential reversibility.”
Traduzione: “I potenziali effetti tossici del prodotto devono essere valutati in relazione agli organi bersaglio, alla dose, alla/e via/e di esposizione, alla durata e alla frequenza dell’esposizione e alla potenziale reversibilità“.
Inoltre, i parametri da osservare sono spiegati a pag. 47:
„Toxicity studies should address the potential of the product for causing local inflammatory reactions, and possible effects on the draining lymph nodes, systemic toxicity and on the immune system. A broad spectrum of information should be obtained from the toxicity studies….”
Traduzione:
“Gli studi di tossicità devono riguardare il potenziale del prodotto di causare reazioni infiammatorie locali e i possibili effetti sui linfonodi drenanti, la tossicità sistemica e il sistema immunitario. Dagli studi di tossicità si deve ottenere un ampio spettro di informazioni….”.
Riguardo a
- Tossicità per lo sviluppo
- Genotossicità,
- Cancerogenicità e
- Farmacocinetica
la linea guida afferma che questi esami “non sono normalmente necessari“.
Tuttavia, dato l’ambito di applicazione della linea guida come definito nella sezione 1.1, le iniezioni di mRNA NON rientrano nell’ambito “normale” della linea guida, per cui il richiedente non può fare affidamento sulle eccezioni definite nella linea guida dell’OMS. Inoltre, la linea guida sottolinea che la necessità di indagini corrispondenti deve essere valutata nel singolo caso del rispettivo prodotto (pagg. 44, 49, 51).
Inoltre, si deve tenere conto del fatto che, nonostante l’eventuale esonero dagli studi da presentare per la sostanza in esame, nel caso di nuovi eccipienti o coadiuvanti, devono essere presentati studi corrispondenti per questi componenti (pag. 51 e segg.).
Sono inoltre essenziali le affermazioni di questa linea guida sulle considerazioni speciali per alcuni tipi di vaccini (p. 54 f). In questi casi, possono essere necessarie ulteriori indagini oltre a quelle descritte nella linea guida. Questa sezione tratta specificamente, tra gli altri, i vaccini a DNA. Si fa riferimento ad altre linee guida dell’OMS. La linea guida dell’OMS sui vaccini a DNA è esplicitamente citata (Doc. A. 23). Secondo questa linea guida, oltre agli esami richiesti in linea di principio, sono necessari anche i seguenti esami:
- Biodistribuzione
- Persistenza e
Si legge a pag. 78:
„The duration and sites of expression of the encoded proteins over time should be investigated. If the encoded protein product is expected to persist for a considerable length of time, the impact of this should be addressed.”
Traduzione:
“La durata e i siti di espressione delle proteine codificate nel tempo devono essere studiati. Se si prevede che il prodotto proteico codificato persista per un periodo di tempo considerevole, si deve considerare l’impatto di questo fatto”.
E a pag. 79 si parla di tossicità per lo sviluppo:
„Integration into reproductive tissue may result in germline alteration. The possibility of distribution to, integration or expression in germline cells must be investigated unless otherwise justified.”
Traduzione:
“L’integrazione nel tessuto riproduttivo può comportare un’alterazione germinale. La possibilità di distribuzione, integrazione o espressione nelle cellule germinali deve essere studiata, a meno che non sia altrimenti giustificato.”
Quest’ultimo significa che devono essere eseguiti degli studi sul punto e che si può derogare a questo requisito solo in casi eccezionali e giustificati. Per la deroga, a sua volta, devono essere presentati studi che dimostrino l’assenza di rischio di penetrazione germinale.
Infine, la Linea Guida OMS 2005 contiene nella sua “Appendice” un elenco impressionante di organi che devono essere esaminati nell’ambito di uno studio di tossicità dopo somministrazione multipla, che viene riprodotto qui di seguito come estratto:


Documento concettuale sulle linee guida per i vaccini a DNA
Lo stesso elevato standard di sicurezza risulta anche dal “Concept Paper on guidance for DNA vaccines” del 15.3.2012, EMEA/CHMP/308136/2007 (Doc. A.24).
Questo documento concettuale tratta della necessità di creare una linea guida per i “vaccini a DNA contro le malattie infettive“. Il contesto è l’esclusione dei “vaccini contro le malattie infettive” dalla definizione di medicinali di terapia genica nel 2009. In precedenza, questi “vaccini a DNA contro le malattie infettive” erano coperti anche dalla linea guida “CPMP note for guidance on the quality, preclinical and clinical aspects of gene transfer medicinal products (CPMP/BWP/3088/99)” (doc. A.20). A causa del ritiro, il CHMP ha ritenuto necessario sviluppare una nuova linea guida per questo tipo di medicinali. Il documento concettuale afferma letteralmente che:
“Guidance for DNA vaccines is provided in the CPMP note for guidance on the quality, preclinical and clinical aspects of gene transfer medicinal products (CPMP/BWP/3088/99), which came into effect in 2001 and is currently under revision. However, it is stated in Directive 2009/120/EC that “gene therapy medicinal products shall not include vaccines against infectious diseases”, and although some principles and requirements of gene therapy apply, the guidance provided in the current gene transfer guideline does not address specific aspects relevant for a DNA vaccine against infectious disease.”
(Traduzione: “La guida per i vaccini a DNA è contenuta nella nota CPMP per la guida sugli aspetti qualitativi, preclinici e clinici dei medicinali a trasferimento genico (CPMP/BWP/3088/99), entrata in vigore nel 2001 e attualmente in fase di revisione. Tuttavia, la Direttiva 2009/120/CE stabilisce che “i medicinali per la terapia genica non comprendono i vaccini contro le malattie infettive” e, sebbene si applichino alcuni principi e requisiti della terapia genica, la guida fornita nell’attuale linea guida sul trasferimento genico non affronta gli aspetti specifici rilevanti per un vaccino a DNA contro le malattie infettive”. (Traduzione: le linee guida per i vaccini a DNA sono contenute nella Comunicazione CPMP per la guida sugli aspetti qualitativi, preclinici e clinici dei medicinali a trasferimento genico (CPMP/BWP/3088/99), entrata in vigore nel 2001 e attualmente in fase di revisione. Tuttavia, la Direttiva 2009/120/CE afferma che “i vaccini contro le malattie infettive non sono medicinali per la terapia genica” e, sebbene si applichino alcuni principi e requisiti della terapia genica, le linee guida contenute nell’attuale direttiva sul trasferimento genico non affrontano le specificità rilevanti per un vaccino a DNA contro le malattie infettive).
Da questa linea guida si evince anche che lo speciale Comitato EMA per le Terapie Avanzate deve SEMPRE e COMUNQUE essere coinvolto nella procedura di approvazione dei vaccini basati sull’ingegneria genetica, cosa che, secondo l’esplicita comunicazione dell’EMA, non è avvenuta nel caso di COMIRNATY!
La linea guida a cui si fa riferimento in questo concept paper, “CPMP note for guidance on the quality, preclinical and clinical aspects of gene transfer medicinal products (CPMP/BWP/3088/99)”, stabilisce elevati requisiti di sicurezza per i vaccini a DNA contro le malattie infettive. Tra le altre cose, è stato necessario condurre studi per dimostrare in quali siti / in quali cellule vengono prodotte le proteine, per quanto tempo vengono prodotte, se esiste una potenziale reattività incrociata, quali effetti immunomodulatori vengono innescati.
Nulla di tutto questo è stato fatto per la sostanza COMIRNATY perché, come conferma esplicitamente l’EMA nel suo rapporto di valutazione, ha semplicemente utilizzato come guida la linea guida per i vaccini convenzionali! È una follia, a dir poco!
Gli studi clinici sulla sostanza autorizzata COMIRNATY non sono mai stati portati a termine (si veda il successivo motivo di nullità II), e sono stati avviati senza che le suddette indagini precliniche obbligatorie fossero state eseguite in conformità allo stato delle conoscenze scientifiche prima dell’inizio dello studio clinico!
Studi di tossicità sui ratti e altri studi precedenti alla sperimentazione clinica FIH – commenti nel libro Project Lightspeed
Inoltre, il libro “Project Lightspeed”[17] contiene informazioni sugli studi del tutto insufficienti condotti da BioNTech prima dell’inizio degli studi clinici FIH. Spiegazioni interessanti si trovano in particolare alle pagine 217 e 218 sullo studio di tossicità condotto sui ratti e a pag. 174 f sugli studi in vitro con uno pseudo-virus.
Studio in vitro sull’efficacia degli anticorpi con uno pseudo-virus
Le informazioni sul test in vitro sull’efficacia degli anticorpi formati, che è stato effettuato solo con uno pseudo-virus – e non con il “virus SARS Cov-2” in questione – e che emergono già dal rapporto di valutazione, si trovano a pag. 174 e seguenti del libro. Da queste spiegazioni si evince anche che anche nella piastra di controllo con le cellule delle scimmie vervet precedentemente non vaccinate sono stati trovati i seguenti anticorpi
“Quando si guardava al microscopio, la risoluzione non ci permetteva di vedere la differenza tra 500 e 50 cellule infette per determinare fino a che punto un vaccino fosse efficace“.[18]
Queste osservazioni del libro sono particolarmente interessanti in relazione a quelle contenute nel Rapporto di valutazione (Assessment Report) dell’EMA a pag. 43:
„The relevance of the pseudovirus assay for authentic SARS-CoV-2 was not discussed. For technical reasons, it was not possible to determine a ratio of neutralizing to non-neutralizing antibodies”.
Traduzione:
“La rilevanza del test dello pseudovirus per il SARS-CoV-2 autentico non è stata discussa. Per ragioni tecniche, non è stato possibile determinare un rapporto tra anticorpi neutralizzanti e non neutralizzanti“.
Studio di tossicità nei ratti
I seguenti commenti sullo studio di tossicità condotto sui ratti si trovano alle pagine 217 e seguenti. Claudia Lindemann, dipendente della BioNTech, aveva trovato un passaggio nelle “Linee guida sulla qualità, la sicurezza e l’efficacia dei vaccini contro l’Ebola” dell’OMS che avrebbe consentito,
“…. di iniziare subito dopo il deposito di una relazione intermedia con gli esperimenti della fase I (nota: sperimentazione clinica FIH). Tale rapporto intermedio dovrebbe includere tutti i dati raccolti durante l’osservazione dei roditori e ricavati dai campioni di sangue prelevati dopo le iniezioni….. Ma la parte più lunga degli studi tossicologici, l’esame degli organi accuratamente prelevati e il controllo microscopico di questi campioni, non dovrebbe necessariamente essere completata prima dell’inizio degli studi sull’uomo…..gli esperti dell’autorità federale hanno dato il loro via libera” (p. 218).[19]
Come promemoria, le linee guida citate finora, che riflettono lo stato delle conoscenze scientifiche, richiedono che la distribuzione e gli effetti della sostanza in esame su tutti gli organi siano disponibili prima che la sperimentazione clinica FIH venga iniziata. Inoltre, questi studi devono essere stati eseguiti in due specie animali, roditori e non roditori. (cfr. II.3.3, 4.2, III.2.2, 3).
BioNTech ha quindi avviato la sperimentazione clinica FIH il 20.4.2020,
senza che siano stati esaminati i risultati degli effetti della sostanza in esame sugli organi degli animali in esame.
Sono stati analizzati solo i parametri ematici.
I requisiti dello stato delle conoscenze scientifiche sono definiti dalle rispettive linee guida dell’UE e dell’ICH, che sono state presentate qui sopra e che – come dimostrato in precedenza – sono state violate su larga scala.
Si noti che la conduzione di una sperimentazione clinica – che nel caso di specie è stata peraltro condotta de facto (dal 27 dicembre 2020) su tutta l’ignara popolazione dell’Unione Europea in grossolana violazione di fondamentali disposizioni giuridiche comunitarie e internazionali (si veda a questo proposito il successivo punto III.) – senza l’esistenza degli studi farmacologico-tossicologici richiesti dallo stato delle conoscenze scientifiche, costituisce un illecito penale.”[20]
Sulla base di quanto sopra, anche l’attuale autorizzazione all’immissione in commercio di COMIRNATY deve essere dichiarata gravemente contraria al diritto dell’UE e quindi nulla.
II° MOTIVO DI NULLITÁ
Grave violazione degli artt. 168 e 169 TFUE, degli artt. 3, 35 e 38 Carta dell’UE, della direttiva 2001/83/CE artt. 8, 11, 26, 54, 58, 59, 86 e segg, Allegato I, Parte I, Parte III, Parte IV, del Regolamento (CE) n. 726/2004 artt. 3-7, 10a, 12, 14, 14a, 20, 20a, 25a, 57, 81, 84a, del Regolamento (CE) n. 507/2006 della Commissione artt. 5 e 7
Nonostante l’omissione degli studi più importanti, il 10 ottobre 2022 l’approvazione inizialmente solo condizionata di COMIRNATY (BioNTech) è stata convertita dalla Commissione UE, su raccomandazione del Comitato per i medicinali per uso umano (CHMP) dell’EMA, in un’approvazione non più condizionata, o autorizzazione senza condizioni specifiche!
Con la decisione di esecuzione del 21 dicembre 2020 (Doc. A.5) sono stati imposti a BioNTech obblighi speciali per l’autorizzazione condizionata di COMIRNATY ai sensi dell’art. 14-a del Regolamento (CE) n. 726/2004, che, insieme ai termini di attuazione, sono stabiliti nelle condizioni per l’autorizzazione condizionata concessa anche sulla base del Regolamento (CE) n. 507/2006 della Commissione.
Nell’ambito degli obblighi specifici di cui al paragrafo 4, il titolare di un’autorizzazione all’immissione in commercio rilasciata in conformità all’articolo 14-a è tenuto a completare gli studi in corso o ad avviarne di nuovi per confermare il rapporto positivo tra rischi e benefici.
BioNTech avrebbe dovuto completare i seguenti studi con un risultato positivo in base all’ultimo stato dell’autorizzazione all’immissione in commercio condizionata prima della semplice conversione assolutamente illegale in un’autorizzazione all’immissione in commercio senza condizioni specifiche (cfr. allegato II, punto E, della decisione di esecuzione della Commissione del 16.9.2022 sulla variazione dell’autorizzazione all’immissione in commercio condizionata rilasciata con decisione C(2020) 9598(finale) per il medicinale per uso umano “Comirnaty-Tozinameran” (doc. A.9 e A.10):
- OBBLIGO SPECIFICO DI COMPLETARE LE ATTIVITÀ POST-AUTORIZZATIVE PER L’AUTORIZZAZIONE ALL’IMMISSIONE IN COMMERCIO SUBORDINATA A CONDIZIONI La presente autorizzazione all’immissione in commercio è subordinata a condizioni; pertanto ai sensi dell’articolo 14-bis del Regolamento 726/2004/CE e successive modifiche, il titolare dell’autorizzazione all’immissione in commercio deve completare, entro la tempistica stabilita, le seguenti attività:
Descrizione Tempistica
Per confermare l’efficacia e la sicurezza di Comirnaty, il dicembre 2023
titolare dell’autorizzazione all’immissione in commercio deve fornire
la relazione finale sullo studio clinico relativa allo studio C4591001
randomizzato, controllato verso placebo, in cieco per l’osservatore.
Per confermare l’efficacia e la sicurezza di Comirnaty, il titolare luglio 2024
dell’autorizzazione all’immissione in commercio deve fornire la
relazione finale sullo studio clinico relativa allo studio C4591007
randomizzato, controllato verso placebo, in cieco per l’osservatore.
Ai sensi dell’art. 14-a, punto (3), le autorizzazioni condizionate possono essere concesse “solo se il rapporto rischi/benefici è positivo e se il richiedente è in grado di fornire dati completi“.
Solo se sono soddisfatti gli obblighi specifici di cui all’articolo 14-bis, paragrafo 4, del regolamento (CE) n. 726/2004, la Commissione, su richiesta del titolare dell’autorizzazione all’immissione in commercio e previo parere favorevole dell’Agenzia[21], può rilasciare un’autorizzazione valida per cinque anni e rinnovabile ai sensi dell’articolo 14, paragrafi 2 e 3.
Se l’EMA conclude che il titolare di un’autorizzazione rilasciata in conformità all’articolo 14-bis del Regolamento (CE) n. 726/2004 non si è conformato agli obblighi previsti dall’autorizzazione, ne informa la Commissione. La Commissione deve quindi adottare una decisione di modifica, sospensione o revoca dell’autorizzazione secondo la procedura di cui all’articolo 10 del regolamento (CE) n. 726/2004.
Nel 2021 si è saputo che la BioNTech (Doc. A. 26), nonostante l’autorizzazione solo condizionata, aveva sciolto i gruppi di controllo dei suoi studi clinici, ai quali era stato somministrato solo placebo. La ragione addotta per lo scioglimento del gruppo di controllo era che sarebbe stato eticamente problematico (Doc. A.27) non somministrare il vaccino ai non vaccinati.
Anche in questo caso, l’audacia con cui l’intera popolazione dell’UE viene esposta a un esperimento genetico illegale non può essere superata. Perché rilasciare per uso generale un preparato che non è stato sistematicamente testato rispetto al gruppo di controllo per verificarne l’effettiva efficacia e soprattutto la sicurezza, viola i più elementari principi del diritto farmaceutico e dei diritti umani! Si veda anche il punto III.
Il Comitato CHMP[22] dell’EMA conferma esplicitamente questo operato, che viola i requisiti dell’autorizzazione all’immissione in commercio, nella sua valutazione ufficiale della richiesta, presentata da BioNTech, di conversione dell’autorizzazione all’immissione in commercio condizionata in autorizzazione all’immissione in commercio regolare[23] (Doc. A.18). La Commissione riconosce inoltre che, a causa dell’eliminazione del gruppo di controllo, la prosecuzione dello studio è diventatainutile, poiché non ci si può aspettare un ulteriore aumento delle conoscenze sull’efficacia e la sicurezza del prodotto. Eliminando le tracce su larga scala, l’industria farmaceutica e le autorità non solo ostacolano efficacemente il chiarimento scientifico, ma proseguono definitivamente l’esperimento di ingegneria genetica condotto su tutta la popolazione dell’UE dal 2021, con la soppressione e la più fondamentale violazione dell’intero quadro giuridico!
Invece di sanzionare immediatamente il produttore a metà del 2021, ai sensi dell’art. 20-bis del Regolamento n. 726/2004/CE, e di modificare, sospendere o revocare l’autorizzazione condizionata, non è successo nulla.
Al contrario, nella decisione di esecuzione del 10 ottobre 2022 (Doc. A.1), la Commissione ha addirittura concesso la regolare autorizzazione all’immissione in commercio, affermando – nonostante lo scioglimento del gruppo placebo già all’inizio del 2021, e quindi nonostante la certa impossibilità di soddisfare le condizioni imposte – che le condizioni specifiche dell’autorizzazione all’immissione in commercio condizionata sarebbero state soddisfatte alla luce dei dati presentati da BioNTech.
Un’autorità di autorizzazione non può agire in modo più impertinente e irresponsabile e criminale! E questo dovrebbe portare a immediate indagini penali in tutta Europa, in particolare presso le sedi dell’EMA, della Commissione UE e del produttore, perché in questo caso sono a rischio la salute e persino la vita dell’intera popolazione europea!
La Commissione ha violato le disposizioni di legge, in particolare l’art. 14-bis comma 8 del Regolamento n. 726/2004/CE e l’art. 7 del Regolamento n. 507/2006/CE della Commissione. Questi stabiliscono che un’autorizzazione all’immissione in commercio condizionata può essere convertita in un’autorizzazione all’immissione in commercio regolare solo quando il produttore ha soddisfatto tutte le condizioni imposte dall’autorizzazione all’immissione in commercio condizionata. Ad esempio, la condizione originaria era quella di continuare gli studi clinici controllati con placebo e di presentarne i risultati nel 2023 o 2024.
“Gli studi pluriennali controllati con placebo (Doc.A.28) sono il “gold standard” per le autorità regolatorie di tutto il mondo per dimostrare l’efficacia e la sicurezza (a lungo termine) dei medicinali. In assenza di tali validi studi, la regolare autorizzazione all’immissione in commercio dei medicinali deve essere obbligatoriamente rifiutata ai sensi dell’art. 12 (1) del Regolamento (CE) n. 726/2004.
Lo scioglimento del gruppo di controllo ha chiaramente violato il presupposto/requisito per l’autorizzazione. I produttori non sono incentivati a condurre studi volontari a lungo termine. I dati a lungo termine sulla sicurezza dei vaccini a mRNA non possono più essere raccolti in gruppi di controllo. L’EMA non ha avviato studi completi in doppio cieco e controllati con placebo.
I dati osservativi derivanti dalla somministrazione per miliardi di volte dei preparati di mRNA non possono sostituire uno studio rigoroso e controllato con placebo. Ciò è tanto più vero nel caso di una così scarsa raccolta e valutazione dei dati sui possibili danni da vaccino, come quella che stiamo vivendo.
L’influenza delle lobby nel processo di autorizzazione ha portato all’annullamento delle regole fondamentali del diritto farmacologico/medico: se si vaccinano persone sane, sono necessari standard di sicurezza più elevati rispetto al caso in cui si sottopongono persone gravemente malate a una sperimentazione di trattamento con farmaci di terapia genica”.[24]
Anche per quanto sopra esposto, l’autorizzazione all’immissione in commercio del COMIRNATY attualmente in vigore è gravemente illegittima ai sensi del diritto dell’UE, e pertanto la decisione di esecuzione della Commissione del 10.10.2022 che concede l’autorizzazione all’immissione in commercio del medicinale per uso umano COMIRNATY, qui contestata, deve essere annullata.
III° MOTIVO DI NULLITÁ
Violazione del Regolamento (UE) n. 536/2014 del Parlamento europeo e del Consiglio del 16 aprile 2014 sulla sperimentazione clinica di medicinali per uso umano
Di fatto si abusa dei cittadini dell’UE, fino ai più piccoli (e ai non nati), come cavie per una sostanza sperimentale somministrata illegalmente. Dal 2021 è in atto un esperimento farmacologico-genetico illegale e criminale condotto sull’intera popolazione dell’UE.
Il considerando (27) del Regolamento (UE) n. 536/2014 recita: “La dignità umana e il diritto all’integrità della persona trovano riconoscimento nella Carta dei diritti fondamentali dell’Unione europea (la «Carta»). In particolare, secondo la Carta nessun intervento nell’ambito della medicina e della biologia può essere eseguito senza il consenso libero e informato della persona interessata. La direttiva 2001/20/CE contiene un ampio complesso di norme per la tutela dei soggetti. Tali norme dovrebbero essere mantenute.”
Il considerando (30) del regolamento recita: “Conformemente agli orientamenti internazionali, il consenso informato dei soggetti dovrebbe essere rilasciato per iscritto… Prima dell’acquisizione del consenso informato il potenziale soggetto dovrebbe ricevere informazioni nel corso di un colloquio preliminare tenuto con un linguaggio di facile comprensione per lo stesso. È opportuno dare al soggetto la possibilità di formulare domande in qualunque momento …”
Secondo l’art. 3 del Regolamento, “Una sperimentazione clinica può essere condotta esclusivamente se:
- a) i diritti, la sicurezza, la dignità e il benessere dei soggetti sono tutelati e prevalgono su tutti gli altri interessi …;
Secondo l’art. 4 del Regolamento, “Una sperimentazione clinica è soggetta a una revisione scientifica ed etica e deve essere autorizzata secondo quanto previsto dal presente regolamento.”
Norme particolarmente severe si applicano ai minori e ad altri gruppi vulnerabili (art. 10 del Regolamento).
Il capo V (Protezione dei soggetti e consenso informato) dell’articolo 28 (Disposizioni generali) del regolamento stabilisce che una sperimentazione clinica è consentita esclusivamente se tutte le seguenti condizioni sono soddisfatte: …
- b) i soggetti … sono stati informati conformemente all’articolo 29, paragrafi da 2 a 6; c) i soggetti … hanno fornito il proprio consenso informato conformemente all’articolo 29, paragrafi 1, 7 e 8;
- d) sono rispettati il diritto all’integrità fisica e mentale dei soggetti …;
- Il consenso informato deve essere dato, datato e firmato per iscritto ………
- Le informazioni fornite al soggetto … al fine di ottenere il consenso informato devono
- consentire al soggetto o al suo rappresentante legalmente designato di comprendere,
- quali sono la natura, gli obiettivi, i benefici, le implicazioni, i rischi e gli inconvenienti della sperimentazione clinica;…
- essere esaurienti, concisi, chiari, pertinenti e comprensibili per i non addetti ai lavori;
- essere comunicati nell’ambito di un precedente colloquio condotto da un membro del team di audit che sia … adeguatamente qualificato;
- contenere informazioni sulla procedura applicabile per il risarcimento dei danni di cui all’articolo 76, paragrafo 1; e
- Includere il numero della sperimentazione UE e le informazioni sulla disponibilità dei risultati della sperimentazione clinica in conformità al paragrafo 6.
- Le informazioni di cui al paragrafo 2 sono registrate per iscritto e messe a disposizione del soggetto. …
Inoltre, norme più severe previste dall’art. 32 del Regolamento si applicano alle sperimentazioni cliniche che coinvolgono i minori.
La popolazione dell’UE, come i due figli minorenni dell’attore, è stata sottoposta a un esperimento di massa da parte della Commissione UE e dell’EMA come cavie per sostanze sperimentali basate sull’ingegneria genetica.
La popolazione dell’UE è stata e continua ad essere ingannata sul fatto che
- i cosiddetti “vaccini” Covid-19 a base di mRNA, come COMIRNATY, sono sostanze che, per composizione e modalità d’azione, corrispondono a medicinali di terapia genica e quindi a medicinali di terapia avanzata,
- non sono stati fatti nemmeno gli studi previsti per le “vaccinazioni” basate sull’ingegneria genetica,
- gli studi clinici inizialmente previsti sono stati semplicemente abbandonati dopo poco tempo (perché i risultati avrebbero mostrato un quadro disastroso sia in termini di mancanza di effetti che di disastroso profilo di sicurezza) e quindi l’effetto e la sicurezza di questa sostanza non sono mai stati dimostrati,
- non esiste una farmacovigilanza adeguata alle proprietà e alle modalità d’azione di questa sostanza, e quindi i dati raccolti sugli effetti collaterali (soprattutto quelli più gravi, come i decessi) sono drammaticamente sottostimati
- eppure, i 13.706 decessi e un totale di 1.214.603 effetti collaterali (per lo più gravi e irreversibili) (Doc. 29) emergenti al 18.2.2023 dal database ufficiale degli effetti collaterali segnalati (EudraVigilance www.adrreports.eu) per il solo COMIRNATY non avrebbero mai dovuto portare a un’autorizzazione all’immissione in commercio, tanto meno a una autorizzazione ordinaria.
L’applicazione di massa di questa sostanza sperimentale, anche ai bambini, con l’inganno, è una grave violazione del Codice di Norimberga, perché solo chi è correttamente e pienamente informato può prendere una decisione “libera”. La popolazione deliberatamente ingannata non è stata quindi in grado di prendere una decisione “libera”, e tutte le “dichiarazioni di consenso” firmate dai vaccinati sono nulli.
Inoltre, negli Stati membri dell’UE, come l’Italia, è stato introdotto un “obbligo di vaccinazione” Covid 19 di ampia portata che ha di fatto colpito anche la fascia di età dei figli minori dell’attore, dopo che i bambini sono stati esclusi dalle attività sportive e culturali, e quindi da importanti contatti sociali, se non erano “vaccinati”- Covid 19.
Nel caso in questione, l’autorizzazione e quindi l’uso nell’uomo non si basano sulla base legalmente richiesta di risultati di studi completi, come specificato nell’Allegato I “Norme e protocolli analitici, tossico-farmacologici e clinici in materia di prove effettuate sui medicinali” della Direttiva 2001/83/CE.
Come indicato nei motivi I e II, mancano dati di studio essenziali, che avrebbero dovuto essere forniti incondizionatamente nel caso di una normale approvazione di un farmaco. D’altra parte, vi sono gravi errori scientifici e problemi di sicurezza non dichiarati, cosicché, complessivamente, il limite verso la sperimentazione umana con la vaccinazione di massa senza sufficienti risultati di studio è stato assolutamente oltrepassato.
Allo stesso tempo, la Commissione sta perseguendo una politica che stabilisce una vaccinazione di fatto obbligatoria per i cittadini europei, come è già evidente, tra l’altro, dalla Strategia europea sui vaccini del 17.6.2020, COM(2020) 245 definitivo, nonché dall’intero volume di acquisti di decine di miliardi di dosi di vaccino (nella più radicale violazione dell’obbligo di trasparenza da parte della stessa Presidente della Commissione), organizzato anche con messaggi SMS tenuti nascosti. Inoltre, la Commissione europea, insieme all’OMS, che è sotto il controllo dell’industria farmaceutica e del complesso militare, sta forzando l’introduzione del passaporto elettronico di vaccinazione.
La mancanza di informazione ed educazione, come mostrato sopra, in combinazione con il fatto che la Commissione è anche l’autorità che rilascia le autorizzazioni per i vaccini Covid e stabilisce misure legislative che pongono l’onere di vaccinarsi ai singoli cittadini dell’Unione Europea, viola i principi giuridici cogenti del diritto internazionale, noti come ius cogens.
I principi sui requisiti del consenso nelle sperimentazioni cliniche (mediche) della Dichiarazione di Helsinki risalgono al Codice di Norimberga, che ha trovato spazio anche nei reati dello Statuto di Roma della Corte penale internazionale.
Il diritto internazionale non è solo “parte integrante” dell’ordinamento giuridico dell’Unione. Gli atti giuridici della Commissione che violano sistematicamente e collettivamente lo ius cogens sono ipso iure nulli ai sensi dell’art. 53 della Convenzione di Vienna sul diritto dei trattati, riconosciuto dal diritto internazionale consuetudinario (cfr. ulteriori riferimenti in letteratura: Schmalenbach, in: Calliess/Ruffert, EUV/AEUV (Fn. 1), art. 216, n. marginale 50; Tomuschat, in: von der Groeben/Schwarze, EUV/EGV (fn. 10), art. 281, n. marginale 43; in particolare Schmalenbach, in: Europarecht als Mehrebenensystem (fn. 4).),67 (75 ss.)).
Inoltre, l’Accordo tra la Corte penale internazionale e l’Unione europea sulla cooperazione e l’assistenza del 10.4.2006 (GU L 115, pag. 50) stabilisce all’art. 4 che le rispettive disposizioni dello Statuto devono essere osservate per l’UE.
L’esecuzione di esperimenti medici o scientifici su esseri umani in tempo di pace, che violano i principi dell’etica medica, costituiscono una violazione dello Statuto di Roma della Corte penale internazionale, in quanto sono il risultato delle azioni della Commissione o della politica dell’Unione. Ai sensi del reato alternativo di cui all’art. 7 par. 1 lett. k dello Statuto di Roma della Corte penale internazionale, con riferimento al divieto in tempo di guerra di “trattamenti inumani, compresi gli esperimenti biologici”, nonché di “infliggere intenzionalmente grandi sofferenze o gravi danni all’integrità fisica o alla salute” ai sensi dell’art. 8 par. 2 lett. a dello Statuto di Roma della Corte penale internazionale, la commissione deliberata di “altri atti inumani di natura analoga” può essere sanzionata come “crimini contro l’umanità” se l’azione dello Stato o delle istituzioni dell’Unione provoca grandi sofferenze o gravi danni all’integrità fisica.
Anche per questo motivo, le decisioni di attuazione della Commissione UE qui impugnate devono essere dichiarate nulle.
IV° MOTIVO DI NULLITÁ
Invalidità delle decisioni di esecuzione impugnate a causa di
abuso e violazione del REGOLAMENTO (CE) N. 507/2006 della Commissione del 29 marzo 2006 relativo all’autorizzazione all’immissione in commercio condizionata dei medicinali per uso umano che rientrano nel campo di applicazione del regolamento (CE) n. 726/2004 del Parlamento europeo e del Consiglio
È evidente che le condizioni di applicabilità del Regolamento (CE) n. 507/2006 della Commissione sono state create artificialmente per poter, senza che ci siano i necessari presupposti, immettere sul mercato sostanze basate sull’ingegneria, nell’ambito di un’autorizzazione condizionata e in un’applicazione di massa forzata (indirettamente o addirittura direttamente sotto la minaccia di esclusione dal lavoro e dalla vita sociale) a tutta la popolazione ignara.
Ai sensi dell’art. 2 del Regolamento (CE) n. 507/2006, l’autorizzazione condizionata di un medicinale può essere presa in considerazione solo se
-
- il medicinale è destinato al trattamento, alla prevenzione o alla diagnosi medica di malattie gravemente debilitanti o pericolose per la vita;
- medicinali da utilizzare in situazioni di emergenza contro una minaccia per la salute pubblica debitamente identificata dall’OMS o dall’UE ai sensi della decisione n. 2119/98/CE.[25]
La presunta situazione di crisi, la cui proclamazione era necessaria anche per poter solo prendere in considerazione un’autorizzazione condizionata di COMIRNATY & Co., è stata creata da un abuso incredibilmente sfacciato del test RT-qPCR e dall’enorme numero di falsi casi positivi di infezione da SARS-CoV-2 in tal modo creati in tutto il mondo (a partire dalla quarta settimana di gennaio 2020). Senza questo enorme numero creato artificialmente (fino al 97%) di falsi casi positivi, il Regolamento (CE) n. 507/2006 non avrebbe mai potuto essere applicato.
La Prof. Dr. Ulrike Kämmerer conclude la sua perizia sul test RT-qPCR con la seguente conclusione (Doc. A.30): “Significatività dei test RT-qPCR per la rilevazione dell’infettività con il coronavirus SARS-CoV-2
- Alla luce dei problemi e delle limitazioni tecniche qui descritte, la RT-qPCR non è uno strumento diagnostico affidabile (e approvato) per il rilevamento dei virus infettivi (in grado di replicarsi) della SARS-CoV-2.
- Inoltre, il risultato del test RT-qPCR puro è solo un valore di laboratorio che, alla luce degli aspetti descritti, non permette mai di affermare in modo valido la presenza di virus infettivi e può essere utilizzato solo in combinazione con una diagnosi dei sintomi clinici (raccolti da operatori sanitari, in Germania medici) per valutare una possibile infezione virale .
Sintesi:
Per il test di persone asintomatiche e persino sintomatiche sulla base di un tampone nasofaringeo, come viene fatto acriticamente in gran numero e prevalentemente da personale non medico SENZA (cruciale qui: contrariamente a quanto richiesto dall’OMS!) prendere l‘anamnesi e i sintomi delle persone testate, la RT-qPCR utilizzata in qualsiasi forma non è adatta a rilevare l’infezione e soprattutto l’infettività con SARS-CoV-2“.[26]
Inoltre, non c’è mai stata una vera e propria situazione di crisi – a meno che non sia stata causata, come in Italia, da assurde misure che inducono alla morte e sono penalmente rilevanti (prevenzione sistematica del trattamento precoce dei malati a domicilio, divieto dell’uso di farmaci che si sono dimostrati molto efficaci per il trattamento della Coivd-19, come l’ivermectina, l’idrossiclorochina, ecc,) la sistemazione dei malati di Coivd-19 in case di riposo, l’uccisione dei pazienti attraverso un assurdo uso massiccio di ventilatori, ecc.) ha causato – perché il tasso di mortalità da infezione (IFR) era basso in generale, e ancora di più per la popolazione sotto i 70 anni.
Lo scienziato medico più citato al mondo, John Ioannidis, MD, insieme ad altri esperti, mostra quanto segue nello studio Age-stratified infection fatality rate of Covid-19 in the non-ederly informed from pre-vaccinated national grunseroprevalence studies[27] (Doc. A. 31):
Per le persone fino a 19 anni, l’IFR è stato dello 0,0003%, per i 20-29 anni dello 0,011%, per i 30-39 anni dello 0,035%, per i 40-49 anni dello 0,129% e per i 60-69 anni dello 0,501%. Questo dà lo 0,035% per le persone di età compresa tra 0 e 59 anni e lo 0,095% per quelle di età compresa tra 0 e 69 anni.
I risultati “suggeriscono che l’IFR prima della vaccinazione è estremamente basso nelle popolazioni non anziane”. A livello globale, “è quindi probabile che l’IFR sia stato rispettivamente dello 0,03% e dello 0,07% per le persone di età compresa tra 0-59 e 0-69 anni”.
In termini di popolazione totale (compresi gli anziani), l’IFR è equivalente a quella di un’influenza moderata.
Pertanto, in nessun momento esistevano i presupposti fondamentali per un’autorizzazione fondamentale di COMIRNATY e degli altri cosiddetti “vaccini”-Covid-19 nell’ambito di un’autorizzazione condizionata ai sensi del Regolamento (CE) n. 507/2006 della Commissione.
Inoltre, l’art. 4 del Regolamento (CE) n. 507/2006 prevede ulteriori condizioni:
- Un’autorizzazione all’immissione in commercio condizionata può essere rilasciata quando il comitato ritiene che, malgrado non siano stati forniti dati clinici completi in merito alla sicurezza e all’efficacia del medicinale, siano rispettate tutte le seguenti condizioni:
- il rapporto rischio/beneficio del medicinale, quale definito all’art. 1, paragrafo 28-bis della direttiva 2001/83/CE, risulta positivo;
- è probabile che il richiedente possa in seguito fornire i dati clinici completi;
- il medicinale risponde ad esigenze mediche insoddisfatte;
- i benefici per la salute pubblica derivanti dalla disponibilità immediata sul mercato del medicinale in questione superano il rischio inerente al fatto che occorrano ancora dati supplementari.
In merito al punto a): In nessun momento il rapporto rischio/beneficio poteva essere considerato e spiegato come positivo in nessun momento, poiché mancano ancora oggi informazioni essenziali per l’esclusione di rischi molto gravi (si vedano i motivi di nullità I e II). Inoltre, a causa del tasso di mortalità da infezione generalmente basso (paragonabile a quello di un’influenza moderata per la popolazione in generale e di fatto zero per i bambini e gli adolescenti) e in considerazione dei casi di eventi dannosi registrati nella banca dati EudraVigilance (tra cui migliaia di decessi e centinaia di migliaia di altri effetti collaterali irreversibili gravissimi), nonostante la mancanza di una farmacovigilanza attiva, non è assolutamente possibile accertare un rapporto rischio/beneficio positivo!
In merito al punto b): Come indicato nel motivo di nullità II, i gruppi placebo delle poche sperimentazioni cliniche sono stati deliberatamente e pianificatamente annullati pochi mesi dopo l’inizio delle sperimentazioni, offrendo ai membri del gruppo di controllo l’inoculazione di COMIRNATY. In questo modo, è stato deliberatamente reso impossibile anche il soddisfacimento di tale requisito, perché i risultati avrebbero fornito un quadro impressionante, disastrosamente negativo del rapporto beneficio/rischio. Questo operato viola i principi fondamentali del diritto farmaceutico, compreso il Regolamento (UE) n. 536/2014 sulla sperimentazione clinica.
In merito al punto c): È stato dimostrato da migliaia di medici in tutto il mondo che di fatto non c’erano mai esigenze mediche insoddisfatte. I medici che curano i loro pazienti al meglio delle loro conoscenze e della loro coscienza sono stati in grado di curare con successo anche pazienti molto anziani a domicilio, utilizzando farmaci come l’Ivermectina, l’Idrossiclorochina e altri (molto spesso in combinazione) – e questo, nonostante li fossero stati posti enormi ostacoli dai governi e dai loro agenti vicari/complici!
In merito al punto d): Sulla base di quanto sopra, non vi è alcuna prova di un beneficio per la salute pubblica: al contrario. A parte gli enormi rischi ed effetti collaterali già noti (per i quali si deve presumere un’estrema sottostima, soprattutto nel caso dei decessi), i rischi elencati nei motivi di nullità I e II sono difficilmente comprensibili nelle loro dimensioni temute e attualmente assolutamente non escludibili. Potrebbero esserci ancora molti danni fisici (ed economici) individuali, e quindi anche economici e di salute pubblica, per l’intera popolazione dell’UE.
Anche per questi motivi, le decisioni di esecuzione della Commissione impugnate in questa sede devono essere annullate.
V° MOTIVO DI NULLITÁ
Annullamento della decisione di esecuzione impugnata per grave violazione degli articoli 168 e 169 TFUE e degli articoli 3, 35 e 38 della Carta dell’Unione europea.
Sulla base dei fatti e delle circostanze sopra esposti e documentati nel presente ricorso, è evidente che le decisioni di esecuzione della Commissione UE qui contestate (in primo luogo la decisione di esecuzione della Commissione del 10.10.2022 – Doc. A.1), violano gravemente i principi sanciti dal legislatore dell’UE nell’articolo 168 TFUE (salute pubblica). Il legislatore dell’UE ha garantito ai cittadini dell’Unione che nella definizione e nell’attuazione di tutte le politiche e attività dell’Unione deve essere assicurato un elevato livello di protezione della salute. L’azione dell’Unione deve essere diretta al miglioramento della salute pubblica, alla prevenzione delle malattie umane e all’eliminazione delle fonti di pericolo per la salute fisica e mentale.
L’UE deve definire misure per stabilire elevati standard di qualità e sicurezza per i farmaci e i dispositivi medici.
Tutti questi obblighi assunti con l’art. 168 TFUE sono stati gravemente violati sia dalla Commissione europea con le decisioni di esecuzione qui impugnate (in primo luogo la decisione di esecuzione della Commissione del 10.10.2022), sia con la direttiva 2009/120/CE (allegato relativo alla parte IV, punto 2.1, ultima frase), sia dal Parlamento europeo e dal Consiglio con la direttiva 2001/83/CE – allegato I, parte IV, punto 2.1, ultima frase. Questi hanno pertanto portato e portano i figli minori dell’attore (e l’intera popolazione dell’UE) concretamente in una situazione di pericolo per la loro salute e la loro vita.
L’articolo 3 della Carta dell’UE (diritto all’integrità) garantisce a ogni persona nell’UE quanto segue: (1) Ogni persona ha diritto all’integrità fisica e mentale. (2) Nel contesto della medicina e della biologia, devono essere rispettati in particolare: il libero consenso informato della persona interessata, secondo le modalità stabilite dalla legge, …, il divieto di utilizzare il corpo umano e le sue parti in quanto tali a scopo di lucro, ….
L’articolo 35 della Carta dell’UE (protezione della salute) garantisce a tutti i cittadini dell’UE che nella definizione e nell’attuazione di tutte le politiche e attività dell’Unione sia assicurato un livello elevato di protezione della salute umana.
L’articolo 169 del TFUE (protezione dei consumatori) garantisce ai consumatori che, per assicurare un livello elevato di protezione dei consumatori, l’UE contribuisca a proteggere la salute e la sicurezza dei consumatori e a promuovere il loro diritto all’informazione.
E secondo l’art. 38 della Carta dell’UE (protezione dei consumatori), le politiche dell’Unione dovrebbero rappresentare un livello elevato di protezione dei consumatori.
Sulla base di quanto precede, è evidente che, con le decisioni di esecuzione qui impugnate, la Commissione dell’UE ha violato in modo grave anche il diritto fondamentale dell’attore e dei suoi figli minori alla tutela dei consumatori e gli obblighi imposti anche alla Commissione, in particolare ai sensi dell’articolo 169 TFUE.”
Ad evidenziare l’enorme rischio connesso con l’inoculazione di queste sostanze e a spiegare che non si tratta di vaccini tradizionali, ma di un cosiddetto pro-farmaco a base genica è anche il Prof.Dr.med. Marco Cosentino (Ordinario di Farmacologia alla facoltà di medicina dell’Università dell’Insubria) nel suo articolo “Capire la farmacologia dei vaccini COVID-19 a mRNA: stiamo giocando a dadi con la spike?” pubblicato sull’International Journal of Molecular Sciences (doc. 33).
Dalla banca dati ufficiale dell’EMA degli eventi avversi (EudraVigilance; vedi https://www.adrreports.eu/it/index.html) alla data del 08.05.2023[28] risultavano nell’UE migliaia di morti e centinaia di migliaia di altri eventi avversi irreversibili segnalati per un probabile nesso causale con l’inoculazione dei cosiddetti “vaccini”-Covid-19. Va evidenziato che non esiste una farmacovigilanza attiva e che specialmente per quanto riguarda i casi di morte e altri casi di eventi avversi gravissimi, la sottostima ammonta a ca. il 99 per cento, e, dunque, i casi raccolti nella banca dati dell’UE EurdraVigilance costituiscono solo l’1 per cento dei casi realmente capitati. Infatti, anche in Italia bisogna lottare per avere le autopsie e esami peritali condotti da esperti liberi da conflitti di interesse e, come siamo venuti nel frattempo a sapere (vedi la premessa) l’AIFA ha sistematicamente soppresso di dati sugli eventi avversi. Dunque, i dati dell’AIFA sono drammaticamente inattendibili e la sottostima degli eventi avversi sarà effettivamente del 99 per cento!
Due anni fa è uscito uno studio di tre scienziati dell’Istituto Superiore della Sanità che invitano a ricalcolare subito il rapporto rischi/benefici dei cosiddetti “vaccini” Covid-19 alla luce delle evidenze sui gravi effetti collaterali (in primis miocardite e pericardite, malattie autoimmuni come la multiple sclerosi) (doc. 34).
Gli allora vertici dell’Istituto Superiore della Sanità, che hanno violato in modo gravissimo il loro obbligo di tutelare la salute e vita dei cittadini italiani, si sono “scagliati contro” i loro stessi scienziati! Vedi al riguardo l’intervista con il Professore Ordinario di Farmacologia nella Facoltà di Medicina e Chirurgia dell’Università dell’Insubria Dott. Marco Cosentino su LaVerità d.d. 6.2.2022 (doc. 34).
In Italia i vertici delle autorità si scagliano contro quei scienziati, peraltro dipendenti delle autorità pubbliche proposte alla tutela della Sanità Pubblica, che cercano di tutelare la popolazione (in primis i bambini e i giovani).
I responsabili dello Stato Florida, invece, hanno chiesto all’Autorità Regolatoria del Farmaco (FDA) e all’Autorità Centrale della Sanità Pubblica degli USA (CDC) di voler disporre la sospensione totale dell’inoculazione dei cosiddetti “vaccini”-Covid-19, visto l’enorme rischio connesso, inclusa la modifica del genoma umano.
Avv.DDr. Renate Holzeisen
Membro del Consiglio della Provincia Autonoma di Bolzano – Lista VITA
https://www.renate-holzeisen.eu
[1] Zulassungsdesaster (in ital. Disastro di autorizzazione), RA Renè M. Kieselmann, Prof. Dr. Gerd Morgenthaler, Dr. Amrei Müller, Prof.Dr. Günter Reiner, RA Dr. Patrick Riebe, Rain Dr. Brigitte Röhrig, Prof.Dr. Martin Schwab
[2] Tradotto in Italiano: “A quanto pare, gli esperti dell’EMA partivano dal presupposto che l’RNA in generale non influisce sull’integrità del genoma della cellula ospite. La prima eccezione a questa regola è nota dal 1970… difficilmente potrà essere considerata una novità nel 2020″.
[3] Traduzione in lingua italiana “2.1. I vaccini a mRNA sono distribuiti in tutto il corpo e interessano in modo preminente i vasi sanguigni”. L’affermazione secondo cui le nanoparticelle di mRNA/lipidi rimangono nel sito di iniezione è ormai ampiamente nota come una palese falsità. I “vaccini” si diffondono rapidamente dal sito di iniezione ai linfonodi regionali e alla circolazione sanguigna… Inoltre, a differenza della maggior parte dei virus, le nanoparticelle di mRNA dei vaccini possono essere assorbite da qualsiasi tipo di cellula, compresi gli endoteli, che costituiscono lo strato cellulare più interno dei vasi sanguigni…. 2.2 L’espressione della proteina spike nell’organismo è diffusa e duratura. Studi su un modello di vaccino a mRNA hanno dimostrato che le nanoparticelle lipidiche, dopo l’iniezione intramuscolare, entrano rapidamente nel flusso sanguigno. Successivamente si accumulano preferenzialmente in alcuni organi, tra cui il fegato, la milza e le ovaie. … almeno i vasi sanguigni stessi sono esposti al vaccino in ogni organo e in ogni tessuto, da cui dobbiamo aspettarci un’espressione diffusa dell’antigene estraneo... Un’altra considerazione importante è la rapidità con cui l’antigene viene espresso e la durata di questa espressione…. un’espressione piuttosto duratura di spike dopo la vaccinazione con mRNA è stata riportata anche da Röltgen et al, che hanno ancora rilevato la proteina spike nei linfonodi 60 giorni dopo la seconda iniezione, e in questo stesso momento hanno anche mostrato la presenza continua di mRNA codificante lo spike. Analogamente, Magen et al. hanno rilevato una forte espressione della proteina spike e la presenza continua dell’RNA a un mese dalla vaccinazione….”
[4] “4.2 Farmacocinetica dei vaccini a mRNA. Le proprietà delle nanoparticelle lipidiche … esercitano una forte influenza sul loro trasporto e sul loro destino all’interno del corpo umano. 4.2.1 Distribuzione negli organi di vaccini a mRNA modello. … il trasporto delle nanoparticelle lipidiche del vaccino può assomigliare a quello delle lipoproteine … la quantità di particelle lipoproteiche assunte e rigirate varia notevolmente tra le cellule dei diversi organi. I seguenti organi ne assorbono quantità particolarmente elevate:
- Il fegato ha un ruolo centrale nel metabolismo delle lipoproteine…
- Le ghiandole endocrine che producono ormoni steroidei … Sono i testicoli, le ovaie e le ghiandole surrenali,
- La placenta ha bisogno di lipoproteine sia per rifornire il feto sia per la sua produzione di ormoni progestinici, necessari per sostenere la gravidanza,
- Le ghiandole mammarie in fase di allattamento acquisiscono grassi e colesterolo dalle lipoproteine e li riconfezionano per rilasciarli nel latte materno.
Con questo in mente, possiamo comprendere alcune delle osservazioni sulla distribuzione dei vaccini a base di mRNA all’interno dell’organismo…Moderna, secondo il rapporto dell’EMA su questo vaccino, … ha presentato alcuni dati sugli animali su un modello di vaccino… In questo studio, sono stati misurati i livelli di mRNA piuttosto che quelli dei lipidi. I risultati dello studio di Moderna sono descritti in modo incompleto nel rapporto, ma a pagina 47 si legge:
Concentrazioni di mRNA più elevate (rispetto ai livelli plasmatici) sono state riscontrate nella milza e nell’occhio. … Bassi livelli di mRNA sono stati rilevati in tutti i tessuti esaminati, tranne il rene. In questo studio è evidente anche la distribuzione epatica dell’mRNA-1647, in linea con quanto riportato in letteratura, secondo cui il fegato è un organo bersaglio comune delle LNP”. …
indipendentemente dal tessuto di un organo specifico, almeno i vasi sanguigni e i loro endoteli saranno esposti alle particelle del vaccino in ogni organo. Di conseguenza, è probabile che vasculiti ed eventi tromboembolici si verifichino in tutti gli organi. Ci si potrebbe aspettare che ulteriori patologie tessuto-specifiche si concentrino sugli organi con alti livelli di accumulo. Tuttavia, come vedremo in seguito, i risultati di questi studi sugli animali probabilmente non forniscono un quadro completo della distribuzione del vaccino mRNA nella pratica. 4.2.2 Correlazione tra la distribuzione negli organi del vaccino modello e i risultati istopatologici … abbiamo riscontrato prove di infiammazione e di espressione della proteina spike indotta dal vaccino nel muscolo cardiaco … e nel cervello …, anche se questi organi hanno accumulato solo livelli relativamente bassi o moderati del vaccino modello negli esperimenti animali di Pfizer e Moderna. L’infiammazione osservata è particolarmente notevole per quanto riguarda il cervello, che dovrebbe essere protetto dalla barriera emato-encefalica. In questo contesto, dobbiamo notare due importanti avvertenze: 1. la barriera emato-encefalica si rompe quando il tessuto cerebrale è colpito da infiammazione. Di conseguenza, la vasculite cerebrale indotta dalla prima iniezione di un vaccino a mRNA potrebbe ammorbidire la barriera emato-encefalica e facilitare l’ingresso delle particelle di vaccino somministrate con una successiva iniezione di richiamo. Sarebbe stato quindi importante esaminare la distribuzione del vaccino negli organi non solo dopo la prima iniezione, ma anche dopo una o più iniezioni ripetute. Tuttavia, questo non è stato fatto negli studi sugli animali di Pfizer e Moderna.
- La proteina spike del SARS-CoV-2 ha dimostrato in diversi studi di compromettere l’integrità della barriera emato-encefalica… La proteina spike, che può essere espressa altrove ma raggiunge il cervello attraverso il flusso sanguigno, può facilitare la penetrazione delle particelle del vaccino nel cervello…. Queste considerazioni, in combinazione con i risultati istopatologici, suggeriscono fortemente che i vaccini a mRNA si distribuiscono in modo più ampio ed efficace di quanto indichino gli studi molto limitati di Pfizer e Moderna su animali…
4.2.3 Tempo di eliminazione e durata dell’attività. Nella Sezione 4.1.4 abbiamo visto che l’mRNA può separarsi dai lipidi dopo l’assorbimento cellulare delle nanoparticelle del vaccino. L’eliminazione di entrambi gli ingredienti deve quindi essere considerata separatamente.
4.2.3.1 Corso temporale dell’eliminazione dell’mRNA. … va sottolineato che nessuno di questi studi ha utilizzato l’mRNA impiegato nei vaccini COVID-19 e che tutti gli studi sono stati condotti su roditori. Questi risultati non possono quindi essere applicati direttamente all’attuale produzione di vaccini a mRNA e al loro uso nei pazienti umani. … L’mRNA del vaccino Covid-19 è stato rilevato a 60 giorni dall’iniezione nei linfonodi … e a 30 giorni nel tessuto muscolare di un arto diverso da quello iniettato … La persistenza a lungo termine dell’mRNA del vaccino in campioni di plasma sanguigno di pazienti iniettati è stata recentemente riportata da Fertig et al. … Questi studi sull’uomo dimostrano che gli mRNA del vaccino possono persistere molto più a lungo di quanto suggeriscano gli studi di Pfizer e Moderna sugli animali.
4.2.3.2 Corso temporale dell’eliminazione dei lipidi. …Secondo il rapporto dell’EMA … Moderna non ha presentato dati sull’eliminazione dei due lipidi sintetici contenuti nel suo vaccino Covid-19 mRNA. …Sebbene l’EMA ci rassicuri sul fatto che l’accumulo dei lipidi all’interno dell’organismo è improbabile, dobbiamo notare che, in primo luogo, le informazioni fornite sono del tutto insufficienti per gli standard abituali di sviluppo e approvazione dei farmaci e, in secondo luogo, che l’assenza di accumulo di lipidi non implica l’assenza di tossicità cumulativa“.
[5] Traduzione in Italiano: “4.3 Tossicità delle nanoparticelle lipidiche. … due specie di lipidi sintetici. I lipidi coniugati con PEG sono i meno abbondanti dei due e l’unico meccanismo di danno documentato consiste in reazioni allergiche a questi lipidi. Al contrario, i lipidi cationici rappresentano quasi la metà del totale dei lipidi presenti nelle LNP del vaccino e possono esercitare una vera e propria tossicità, senza alcuna “assistenza” da parte del sistema immunitario adattativo. …
4.3.2 Segnalazione infiammatoria da parte dei lipidi cationici. Diversi studi sperimentali hanno dimostrato che i lipidi cationici simili a quelli utilizzati nei vaccini COVID-19 di Pfizer e Moderna inducono forti reazioni infiammatorie. … Questo concorda con la frequente osservazione di reazioni infiammatorie locali e anche sistemiche tra i riceventi del vaccino COVID-19….”
5 Genotossicità dei vaccini a mRNA … 5.2.1.4 Sintesi. Anche se non era ancora stato dimostrato sperimentalmente quando i vaccini a mRNA COVID-19 sono stati approvati in via d’urgenza, esistevano numerosi precedenti che suggerivano la forte possibilità che copie di DNA dell’mRNA del vaccino si formassero e si inserissero nel genoma cellulare. Invece di ignorare questo rischio come hanno fatto, l’EMA e le altre autorità di regolamentazione avrebbero dovuto obbligare Pifzer e Moderna a condurre gli studi necessari per escludere questo rischio prima di dare il via libera all’autorizzazione… I risultati riportati da Aldén et al., anche se per certi aspetti preliminari, pongono alcuni interrogativi molto seri che non possono più essere ignorati dalle autorità di regolamentazione…. Inattivazione genica. L’inserzione può avvenire all’interno di un gene e interromperlo. Questo può portare alla perdita di importanti prodotti genici cellulari (cioè proteine) e quindi, potenzialmente, allo sviluppo di malattie, tra cui il cancro. … Regolazione genica. I meccanismi di regolazione trascrizionale ed epigenetica possono essere influenzati, modulando così i livelli di espressione delle proteine verso l’alto e verso il basso con risultati imprevedibili e indesiderati. Gli effetti regolatori indiretti possono riguardare anche geni distanti situati su altri cromosomi”… Attivazione di oncogeni… l’insorgenza di neoplasie attraverso l’integrazione del DNA e l’attivazione di geni promotori del cancro (oncogeni) è stata dimostrata in studi clinici … per il trattamento genetico dei bambini …. Queste neoplasie si manifestano in genere solo alcuni anni dopo il completamento del trattamento. Pertanto, indagini approfondite a lungo termine sui possibili effetti genotossici dell’integrazione cromosomica sono assolutamente necessarie, sia in fase preclinica che di sperimentazione clinica, per una valida analisi dei benefici e dei rischi…. Il rischio di inserimento nel DNA cromosomico deve essere preso in seria considerazione…. Malattia autoimmune. L’integrazione del gene della proteina spike nella cellula ospite potrebbe portare all’espressione permanente di questo antigene e quindi indurre una malattia cronica di tipo autoimmune… Integrazione germinale. … Gli esperimenti condotti da Pfizer indicano un alto livello di accumulo del vaccino nelle ovaie … Inoltre, LINE-1 e altri retrotrasposoni sono attivi e causano eventi di inserimento genomico negli ovociti umani … In combinazione, questi risultati indicano che le sequenze geniche di mRNA possono essere integrate nel DNA degli ovociti e quindi nella linea germinale umana. Non si può escludere nemmeno l’inserimento nelle cellule germinali maschili, anche se nello studio animale citato i livelli tissutali del vaccino mRNA modello nei testicoli erano significativamente più bassi rispetto alle ovaie. Se ciò dovesse effettivamente verificarsi – se le cellule germinali degli individui vaccinati dovessero essere rese transgeniche – il rischio di generare o concepire figli transgenici non sarà limitato solo a questi individui, ma sarà necessariamente condiviso dai loro coniugi attuali o futuri. In effetti, un’intera generazione di futuri genitori sarà esposta a questo rischio. … Sintesi. L’integrazione delle sequenze di mRNA nelle cellule somatiche è probabile e comporta un rischio di cancro e di malattie autoimmuni. Inoltre, non si può negare il rischio di integrazione germinale, con conseguente prole transgenica. Questi rischi devono essere affrontati con urgenza attraverso studi approfonditi sugli animali. Nel frattempo, le autorizzazioni di tutti i vaccini a mRNA attualmente in uso devono essere urgentemente revocate“.
[6] Vedi nota al punto 4.2.3. tossicologia a)
[7] Vedi nota al punto 4.2.3. tossicologia a)
- [8] Traduzione in Italiano: ” 3.2.3 . Genotossicità
Non sono stati condotti studi sulla genotossicità, cioè sui danni al materiale genetico umano che potrebbero portare a mutazioni ereditarie e al cancro. Nel rapporto dell’EMA …, ciò viene giustificato come segue:
Non sono stati forniti studi di genotossicità. Ciò è accettabile perché i componenti della formulazione del vaccino sono lipidi e RNA, che non dovrebbero avere un potenziale genotossico. La valutazione del rischio effettuata dal richiedente mostra che il rischio di genotossicità legato a questi eccipienti [cioè i lipidi sintetici] è molto basso sulla base dei dati della letteratura.
In realtà, è noto che le LNP contenute nel BNT162b2 possono entrare in tutti i tipi di cellule – questo è, dopo tutto, lo scopo della loro inclusione in questa preparazione vaccinale. È anche noto che, una volta all’interno della cellula, i lipidi cationici disturbano la funzione mitocondriale (respirazione cellulare) e causano stress ossidativo, che a sua volta porta a danni al DNA.
Va detto che due dei lipidi utilizzati da Pfizer – il lipide cationico ALC-0315 e il lipide pegilato ALC-0159, che rappresentano rispettivamente il 30-50% e il 2-6% del contenuto lipidico totale – non erano stati precedentemente approvati per l’uso nell’uomo. L’atteggiamento cavilloso di Pfizer e dell’EMA nei confronti dell’uso di sostanze chimiche nuove e finora non provate come componenti di preparati per farmaci o vaccini senza studi completi sulla tossicità, compresa la genotossicità, è del tutto antiscientifico e inaccettabile“.
[9] Traduzione in Italiano: “3.1.1.7 Rischi potenziali per la fertilità e per il neonato allattato al seno
Un alto livello di espressione di spike nelle ovaie solleva la prospettiva di un danno significativo a tale organo, con possibili conseguenze sulla fertilità femminile. L’assorbimento del vaccino da parte delle cellule della ghiandola mammaria apre due possibili vie di tossicità per il bambino allattato al seno: in primo luogo, l’espressione della proteina spike e la sua secrezione nel latte materno e, in secondo luogo, il trasferimento del vaccino nel latte. Le ghiandole mammarie sono apocrine, il che significa che si staccano e rilasciano frammenti del loro stesso citoplasma nel latte; pertanto, tutto ciò che ha raggiunto il citoplasma potrebbe anche raggiungere il latte materno. A questo proposito, notiamo che sia il database VAERS che il registro degli eventi avversi ai farmaci dell’UE (EudraVigilance) riportano casi di decesso in neonati allattati al seno dopo la vaccinazione delle loro madri (vedi Sezione 3.1.3.6). ….
- 2.4 Tossicità per la riproduzione
La tossicità riproduttiva è stata valutata utilizzando una sola specie (ratti) e su un numero ridotto di animali (21 cucciolate). È stato rilevato un aumento di oltre due volte della perdita di embrioni prima dell’impianto, con un tasso del 9,77% nel gruppo del vaccino, rispetto al 4,09% nel gruppo di controllo. Invece di limitarsi ad affermare … che il valore più alto era “all’interno dell’intervallo dei dati storici di controllo”, lo studio avrebbe dovuto dichiarare senza ambiguità se questa differenza fosse statisticamente significativa o meno; e se non lo fosse stata, il numero di esperimenti avrebbe dovuto essere aumentato per garantire la potenza statistica richiesta. Lo stesso vale per le osservazioni sulla “bassissima incidenza di gastroschisi, malformazioni della bocca/mascella, arco aortico destro e anomalie delle vertebre cervicali”. Nel complesso, questi studi sono descritti in modo inadeguato e, a quanto pare, sono stati anche condotti in modo inadeguato. “
[10] Traduzione in Italiano: “3.1.1. …Comirnaty, come tutti gli altri vaccini COVID-19 basati sui geni, provoca l’espressione in vivo di una proteina strutturale del SARS-CoV-2, la cosiddetta proteina spike, che si trova naturalmente sulla superficie della particella virale. …. L’idea chiave del vaccino Comirnaty è la seguente:
- un mRNA sintetico che codifica la proteina spike viene complessato con una miscela di lipidi sintetici neutri e cationici (con carica positiva), che si raggruppano in nanoparticelle lipidiche (LNP);
- Dopo l’iniezione, le LNP facilitano l’assorbimento dell’mRNA nelle cellule ospiti, dove l’mRNA causerà l’espressione (sintesi) della proteina spike;
- la proteina spike apparirà sulla superficie delle cellule ospiti e indurrà essa stessa una reazione immunitaria”.
[11] Traduzione in Italiano: “… si prega di notare che nessuno dei vaccini COVID-19 autorizzati, compresi Comirnaty e Spikevax, ha ricevuto input da parte del comitato EMA-S per le terapie avanzate (CAT) e pertanto non esiste alcuna documentazione che dimostri il coinvolgimento del comitato CAT nella rispettiva procedura di autorizzazione all’immissione in commercio condizionata (CMA)”.
“Si noti che i vaccini COVID-19 a base di mRNA non sono una terapia genica. Secondo la definizione di medicinali per la terapia genica di cui all’Allegato I della Direttiva 2001/83/CE, PARTE IV, punto 2.1., sono medicinali per la terapia genica solo quelli utilizzati o somministrati all’uomo allo scopo di regolare, riparare, sostituire, aggiungere o eliminare una sequenza genetica. Non è questo il caso dei vaccini a base di mRNA. Pertanto, il coinvolgimento del CAT non era previsto per nessun vaccino COVID-19”.
[12] Traduzione in Italiano:
” …. Si prega di notare che il richiedente dell’autorizzazione di Comirnaty non ha presentato studi di genotossicità o cancerogenicità, in quanto i componenti della formulazione del vaccino sono lipidi e RNA che non dovrebbero avere un potenziale genotossico. …. Gli studi di genotossicità o di cancerogenicità non sono di solito necessari per la formulazione finale del vaccino e quindi non vengono normalmente richiesti al richiedente. Ciò è in linea con le due linee guida dell’OMS sulla valutazione non clinica dei vaccini (OMS 2005, OMS 2013).”
[13] Traduzione in Italiano:
“Studi di farmacologia della sicurezza
Non sono stati condotti studi di farmacologia di sicurezza con il BNT162b2. Il Richiedente dell’autorizzazione fa riferimento al fatto che sono non ritenuti necessari secondo le linee guida dell’OMS (OMS, 2005). Inoltre, non sono stati registrati esiti su funzioni di organi vitali negli studi di tossicologia a dosi ripetute. Pertanto, l’assenza di studi di farmacologia della sicurezza è approvato dal CHMP”.
[14] Traduzione in Italiano: “La linea guida dell’OMS sulla valutazione non clinica dei vaccini (2) è stata pubblicata nel 2005 ed è il risultato di una collaborazione tra esperti di diverse agenzie regolatorie, agenzie sanitarie, istituzioni accademiche e produttori di vaccini. I regolatori dell’UE hanno partecipato attivamente a questo lavoro. A seguito di discussioni all’interno del CHMP Safety Working Party e del Vaccine Working Party, è stato deciso di eliminare la linea guida del CPMP sui test preclinici farmacologici e tossicologici dei vaccini e di fare riferimento alla linea guida dell’OMS sulla valutazione non clinica dei vaccini“.
[15] “Una linea guida del CHMP non ha valore legale. Come si legge nel documento dell’EMA che descrive lo status delle linee guida scientifiche: “Le linee guida scientifiche devono essere considerate come una posizione comunitaria armonizzata che, se seguita dalle parti interessate come i richiedenti, i titolari di autorizzazione all’immissione in commercio, gli sponsor, i produttori e le autorità di regolamentazione, faciliterà la valutazione, l’approvazione e il controllo dei medicinali nell’Unione Europea. Tuttavia, è possibile adottare approcci alternativi, a condizione che siano adeguatamente giustificati”. (3). Pertanto, facendo riferimento alla linea guida dell’OMS, si applicano gli stessi principi”.
[16] Traduzione in Italiano: “I vaccini per uso umano comprendono uno o più dei seguenti elementi: microrganismi inattivati con mezzi chimici e/o fisici che conservano proprietà immunogene adeguate; microrganismi viventi che sono stati selezionati per la loro attenuazione pur conservando proprietà immunogene; antigeni estratti da microrganismi, secreti da essi o prodotti mediante la tecnologia del DNA ricombinante; microrganismi chimerici; antigeni prodotti in vivo nell’ospite vaccinato in seguito alla somministrazione di un vettore o di un acido nucleico vivo o antigeni prodotti mediante sintesi chimica in vitro. Gli antigeni possono essere allo stato nativo, troncati o modificati in seguito all’introduzione di mutazioni, detossificati con mezzi chimici o fisici e/o aggregati, polimerizzati o coniugati a un vettore per aumentarne l’immunogenicità. Gli antigeni possono essere presentati allo stato puro o insieme a un adiuvante, o in combinazione con altri antigeni, additivi e altri eccipienti”.
[17] Project Lightspeed – The Road to the BioNTech Vaccine – and to a Medicine of Tomorrow, Joe Miller, Uğur Şahin and Özlem Türeci, Rowohlt Verlag, 2. edition, October 2021
[18] Testo originale in lingua tedesca: “„nur eine kleine Zahl von Zellen infiziert (hatte): 500 von 40.000. Beim Blick durch’s Mikroskop erlaubte es die Auflösung nicht, den Unterschied zwischen 500 und 50 infizierten Zellen zu erkennen, um festzustellen, inwieweit ein Impfstoff Wirkung zeigte.“
[19] Testo originale in tedesco: „…. bereits nach Vorlage eines Interimberichts mit den Versuchen von Phase I (Anm. FIH klinische Studie) zu beginnen. Ein solcher vorläufiger Bericht müsste all die Daten enthalten, die bei Beobachtung der Nager erhoben worden waren und die aus den nach den Injektionen entnommenen Blutproben stammten….. Doch der zeitaufwändigste Abschnitt der toxikologischen Studien, die Untersuchung der sorgfältig entnommenen Organe und die mikroskopische Kontrolle dieser Proben, müsste vor Beginn der Humanstudien nicht unbedingt abgeschlossen sein…..die Experten der Bundesbehörde gaben ihr grünes Licht“ (S. 218)
[20] Paragrafi da 85 a 107 e 109 e da 111 a 113 RA Dr Brigitte Röhrig, esperta di diritto dei farmaci
[21] Si intende EMA (Agenzia europea del farmaco)
[22] CHMP = Committee for Medicinal Products for Human Use
[23] Non più sottoposta a specifiche condizioni
[24] René M. Kieselmann (avvocato), Prof. Dr. Gerd Morgenthaler, Dr. Amrei Müller, Prof. Dr. Günter Reiner, Dr. Patrick Riebe (avvocato), Dr. Brigitte Röhrig (avvocato), Prof. Dr. Martin Schwab
[25] Il punto 3 non ha alcun ruolo in questo contesto.
[26] Testo orginale in lingua tedesca: „Aussagekraft der RT-qPCR Tests zur Erkennbarkeit einer Infektiosität mit dem Coronavirus SARS-CoV-2
- Vor dem Hintergrund der hier dargelegten Probleme und technischen Limitationen ist die RT-qPCR kein geeignetes zuverlässiges (und zugelassenes) Diagnostikmittel zum Nachweis von infektiösen (replikationsfähigen) SARS-CoV-2 Viren.
- Ferner ist das reine RT-qPCR Testergebnis nur ein Laborwert, der angesichts der dargelegten Aspekte niemals eine valide Aussage über das Vorhandensein infektiöser Viren erlaubt und nur in Zusammenschau mit einer klinischen Symptomdiagnose (erhoben durch Gesundheitsdienstleister, in Deutschland Mediziner) überhaupt zur Abschätzung einer möglichen Virusinfektion eingesetzt werden darf.
Zusammenfassung:
Zur Testung asymptomatischer und selbst symptomatischer Menschen anhand eines Nasen-Rachenabstrichs, wie er massenweise unkritisch und überwiegend von nicht-medizinischen Personal OHNE (hierbei entscheidend: entgegen der WHO-Forderung!) Anamnese- und Symptomerhebung bei den Getesteten erfolgt, ist die eingesetzte RT-qPCR in jeglicher Form nicht tauglich, eine Infektion und vor allem eine Infektiosität mit SARS-CoV-2 zu erkennen.“
[27] In Italiano: Tasso di mortalità per infezione di Covid-19 stratificato per età nei non informati da studi nazionali di grunseroprevalenza pre-vaccinati
[28] E nel frattempo se ne sono aggiunti, purtroppo, tantissimi altri casi.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Un disastro, i colpevoli devono essere puniti, la giustizia deve fare il suo corso.
Con questa CORROTTA magistratura pensare di ottenere giustizia per i tanti ASSASSINATI e i troppi danneggiati è come guidare su una strada ghiacciata a oltre 100 km all’ora. Inevitabilmente si va a sbattere. Occorre, in primis, avere maggioranza politica per quindi AZZERARE queste ASSOCIAZIONI a DELINQUERE ed istituire uno o più Tribunali di sana pianta POPOLARE dove prevalga la presenza di quella grande maggioranza di popolo che si alza all’alba per andare nei campi o negli allevamenti per PRODURRE ciò che necessità al mantenimento alla vita.
Questo SISTEMA è marcio oltre le fondamenta, NON esiste alcuna possibilità di ripristinare una parvenza di legalità, giustizia e amore/rispetto per il prossimo.
Un caro saluto da Lillo. Calogero.lamantia@alice.it